蛋白质结构预测方法综述
发布时间: 2024-03-01 12:23:00 阅读量: 114 订阅数: 42 
# 1. 蛋白质结构预测概述
蛋白质在生物体内发挥着重要的功能,其结构决定了其功能。蛋白质结构预测即通过一定的方法和技术,尝试推测蛋白质的三维结构。本章将介绍蛋白质结构预测的概念、意义、应用以及所面临的挑战和难点。
## 1.1 什么是蛋白质结构预测
蛋白质结构预测是指利用计算方法尝试推测蛋白质的三维结构,通常包括确定蛋白质的二级结构、三级结构和蛋白质结构中的域。通过蛋白质结构预测,可以更深入地理解蛋白质的功能及其与其他生物分子的相互作用。
## 1.2 蛋白质结构预测的意义和应用
蛋白质结构预测在药物设计、疾病诊断、基因工程等许多领域具有重要意义。准确地预测蛋白质结构可以帮助科学家设计新型药物、理解疾病机制、改造蛋白质功能等。
## 1.3 蛋白质结构预测的挑战和难点
蛋白质结构的预测是一项复杂且具有挑战性的任务。由于蛋白质结构受到多种因素的影响,如氨基酸序列之间的相互作用、水溶性等,因此准确地预测蛋白质的结构仍然是一个难题。此外,蛋白质结构的折叠过程也存在许多不确定性,增加了预测的难度。
在接下来的章节中,我们将介绍蛋白质结构预测的不同方法和技术,以及各种方法的特点和应用。
# 2. 基于序列的蛋白质结构预测方法
蛋白质结构预测是利用已知的蛋白质序列信息,通过一系列计算方法和算法来推断其空间结构的过程。基于蛋白质序列的结构预测方法是其中最为基础和重要的一类,涵盖了多种技术和算法。
### 2.1 基于序列相似性的方法
基于序列相似性的预测方法是通过将待预测蛋白质序列与已知的蛋白质序列进行比对,从中获取结构信息。常用的方法包括Pairwise Sequence Alignment(如Smith-Waterman算法和Needleman-Wunsch算法)、Multiple Sequence Alignment等。
```python
# 以Smith-Waterman算法为例
def smith_waterman(seq1, seq2):
# 实现Smith-Waterman算法的代码
return alignment_score
seq1 = "ATCGTACG"
seq2 = "ATGGTCG"
score = smith_waterman(seq1, seq2)
print("Smith-Waterman算法得分:", score)
```
**总结:** 基于序列相似性的方法通过比对蛋白质序列之间的相似性来推断结构信息。
### 2.2 基于机器学习的方法
机器学习在蛋白质结构预测中发挥着重要作用,通过训练模型来预测蛋白质的结构。常用的机器学习算法有Random Forest、Support Vector Machine(SVM)、Gradient Boosting等。
```java
// 使用SVM算法进行蛋白质结构预测
public class ProteinStructurePrediction {
public static void main(String[] args) {
// SVM算法实现代码
double accuracy = svm_algorithm.train(training_data);
System.out.println("SVM算法准确率:" + accuracy);
}
}
```
**总结:** 机器学习方法通过训练模型来预测蛋白质的结构,可以提高预测的精准度。
### 2.3 基于深度学习的方法
近年来,深度学习技术在蛋白质结构预测领域取得了许多突破,如使用神经网络进行结构预测、利用卷积神经网络(CNN)进行特征提取等。
```python
# 使用深度学习模型进行蛋白质结构预测
import tensorflow as tf
model = tf.keras.Sequential([
tf.keras.layers.Dense(128, activation='relu'),
tf.keras.layers.Dense(64, activation='relu'),
tf.keras.layers.Dense(3, activation='softmax')
])
model.compile(optimizer='adam', loss='sparse_categorical_crossentropy', metrics=['accuracy'])
model.fit(training_data, training_labels, epochs=10)
```
**总结:** 深度学习方法利用神经网络等深度学习技术进行蛋白质结构预测,能够处理更加复杂的结构信息。
# 3. 基于结构的蛋白质结构预测方法
蛋白质的结构对其功能具有至关重要的影响,因此基于蛋白质结构的预测方法在生物信息学领域扮演着重要的角色。本章将介绍基于结构的蛋白质结构预测方法,包括蛋白质结构比对、蛋白质结构建模和分子动力学模拟等内容。
### 3.1 蛋白质结构比对方法
蛋白质结构比对是通过将目标蛋白质的序列或结构与已知蛋白质的序列或结构进行比对,从而推断目标蛋白质的结构特征。常用的蛋白质结构比对方法包括:
**3.1.1 蛋白质序列比对**
蛋白质序列比对是基于蛋白质序列的相似性进行比对,通过寻找具有相似氨基酸序列的蛋白质来推断目标蛋白质的结构特征。常用的序列比对工具包括BLAST、PSI-BLAST等。
**3.1.2 结构比对算法**
结构比对算法主要通过比较蛋白质的三维结构来推断它们之间的关系。一些常用的结构比对算法包括TM-align、DALI、CE等,通过计算结构之间的相似性得分来评估它们的结构相似程度。
### 3.2 蛋白质结构建模方法
蛋白质结构建模是通过已知的蛋白质结构或结构片段来预测目标蛋白质的结构。常用的蛋白质结构建模方法包括:
**3.2.1 蛋白质同源建模**
蛋白质同源建模是利用与目标蛋白质序列相似度较高的已知结构作为模板来进行蛋白质结构的预测。常用的同源建模软件包括MODELLER、SWISS-MODEL等。
**3.2.2 蛋白质碎片拼装**
蛋白质碎片拼装是将已知结构中的片段与目标蛋白质的序列进行匹配,然后进行蛋白质结构的组装。这种方法常用于预测蛋白质的局部结构。
### 3.3 分子动力学模拟方法
分子动力学模拟是一种基于物理化学原理的方法,通过模拟蛋白质分子在原子水平上的运动来预测其结构和功能。分子动力学模拟方法在蛋白质结构预测中发挥着重要作用,能够模拟蛋白质在不同环境条件下的构象变化和相互作用。
以上介绍了基于结构的蛋白质结构预测方法,这些方法在不同场景下具有各自的优势和适用性,可以根据具体问题的需求选择合适的方法进行蛋白质结构预测。
# 4. 整合方法及进展
蛋白质结构预测的研究领域涉及多种方法和算法,为了提高预测的准确性和可靠性,研究者们开始尝试将不同的方法进行整合,以期取长补短,取得更好的效果。本章将介绍蛋白质结构预测领域常见的整合方法及最新进展。
#### 4.1 融合序列和结构信息的方法
在蛋白质结构预测中,序列信息和结构信息都包含着重要的特征。为了充分利用两者的优势,研究者们提出了一系列融合序列和结构信息的方法。这些方法通常通过构建融合模型,在训练和预测过程中同时考虑序列和结构信息,从而取得更好的效果。
#### 4.2 综合多种算法的整合方法
除了融合序列和结构信息外,还有一种常见的整合方法是综合多种不同的算法。由于不同算法在不同数据集或任务上表现优势各不相同,综合多种算法的方法可以提高蛋白质结构预测的鲁棒性和泛化能力。研究者们通过集成学习、模型融合等技术,将多种算法结合起来,取得更加可靠的结果。
#### 4.3 蛋白质结构预测领域的最新进展
随着人工智能和深度学习技术的发展,蛋白质结构预测领域也迎来了新的突破和进展。越来越多的研究者开始探索将深度学习应用于蛋白质结构预测中,利用神经网络等技术来提取更丰富的特征信息,进一步提高预测的精确性和速度。未来,随着技术的不断进步和研究的深入,蛋白质结构预测领域将迎来更多的创新和突破。
# 5. 蛋白质结构预测工具和软件介绍
蛋白质结构预测工具和软件是在蛋白质结构预测领域中至关重要的辅助工具,能够帮助科研人员快速准确地进行蛋白质结构的预测和分析。本章将介绍一些常用的蛋白质结构预测工具、它们的功能特点以及比较分析,并通过一些使用案例展示它们的具体应用。
#### 5.1 常用的蛋白质结构预测工具
在蛋白质结构预测领域,有许多著名的工具和软件被广泛应用,其中包括:
- **SWISS-MODEL**:SWISS-MODEL是一种常用的蛋白质结构建模工具,通过基于比对的方式进行蛋白质结构的预测和建模。
- **I-TASSER**:I-TASSER是一种综合了多种算法的蛋白质结构预测工具,能够进行蛋白质的全原子模型构建和功能预测。
- **ROSETTA**:ROSETTA是一种以蛋白质二级结构信息为输入的蛋白质结构预测软件,采用蛋白质的能量函数进行构象搜索和优化。
#### 5.2 软件功能与比较分析
这些工具各有其独特的特点和功能,比如SWISS-MODEL在蛋白质结构建模领域有着较高的准确性,I-TASSER能够整合多种算法提高预测的覆盖范围,而ROSETTA则利用蛋白质的物理化学性质进行结构构建。
在进行软件的选择时,需根据具体需求和所研究的蛋白质类型来选择最适合的工具,同时也可以结合多种软件进行综合应用,以提高蛋白质结构预测的准确性和可靠性。
#### 5.3 使用案例展示
以下为一个使用SWISS-MODEL进行蛋白质结构建模的简单Python示例:
```python
from modeller import *
from modeller.automodel import *
env = environ()
a = automodel(env, alnfile='alignment.ali', knowns='template', sequence='query')
a.starting_model = 1
a.ending_model = 5
a.make()
```
上述代码片段展示了使用SWISS-MODEL进行蛋白质结构建模的过程,通过输入alignment文件和已知结构的模板,即可生成预测的蛋白质结构模型。通过这样的使用案例,可以更直观地了解这些工具在蛋白质结构预测中的应用方式和效果。
# 6. 蛋白质结构预测方法的未来发展方向
在蛋白质结构预测领域,随着科学技术的不断进步和数据量的不断增加,未来的发展方向将更加注重以下几个方面:
### 6.1 深度学习在蛋白质结构预测中的应用
随着深度学习技术在各个领域的成功应用,越来越多的研究者开始探索如何将深度学习应用于蛋白质结构预测中。通过构建更加复杂的神经网络模型,可以更好地捕捉蛋白质结构中的关键特征,从而提高预测的准确性和效率。目前,基于深度学习的方法已经取得了一些令人瞩目的成果,但在模型解释性和数据需求方面仍存在挑战,未来的研究将重点关注这些问题。
```python
# 以PyTorch为例,展示深度学习在蛋白质结构预测中的应用示例代码
import torch
import torch.nn as nn
import torch.optim as optim
# 构建一个简单的蛋白质结构预测模型
class ProteinPredictor(nn.Module):
def __init__(self):
super(ProteinPredictor, self).__init__()
self.fc1 = nn.Linear(100, 64)
self.fc2 = nn.Linear(64, 32)
self.fc3 = nn.Linear(32, 3) # 假设预测三维坐标
def forward(self, x):
x = torch.relu(self.fc1(x))
x = torch.relu(self.fc2(x))
x = self.fc3(x)
return x
# 实例化模型和定义优化器
model = ProteinPredictor()
optimizer = optim.Adam(model.parameters(), lr=0.001)
# 准备训练数据并进行模型训练
# 模型预测示例
input_data = torch.tensor([0.1, 0.2, ..., 0.9]) # 输入蛋白质序列特征
output = model(input_data)
print(output)
```
### 6.2 结合实验数据的精准预测方法
除了基于计算的方法外,未来的发展将更多地结合实验数据,利用实验结果对蛋白质结构进行验证和修正。通过结合实验数据,可以提高预测模型的精准度,并使预测结果更加可靠。因此,未来的研究将更加注重如何有效地整合实验数据和计算模型,实现蛋白质结构预测的精准化和可靠化。
### 6.3 个性化蛋白质结构预测与医疗应用
随着个性化医疗的发展,蛋白质结构预测也将朝着个性化方向发展。个体之间蛋白质结构的差异对于疾病的发生和治疗有着重要影响,因此个性化蛋白质结构预测将有助于设计针对性更强的药物和治疗方案。未来的研究将探索如何通过个性化分析蛋白质结构,实现更加精准的健康管理和医疗应用。
以上是蛋白质结构预测方法未来发展方向的简要介绍,随着科技的不断进步和研究的深入,相信蛋白质结构预测领域将迎来更加辉煌的未来。

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
Adblock Plus高级应用:如何利用过滤器提升网页加载速度

# 摘要
本文全面介绍了Adblock Plus作为一款流行的广告拦截工具,从其基本功能到高级过滤策略,以及社区支持和未来的发展方向进行了详细探讨。首先,文章概述了Adb
【QCA Wi-Fi源代码优化指南】:性能与稳定性提升的黄金法则

# 摘要
本文对QCA Wi-Fi源代码优化进行了全面的概述,旨在提升Wi-Fi性能和稳定性。通过对QCA Wi-Fi源代码的结构、核心算法和数据结构进行深入分析,明确了性能优化的关键点。文章详细探讨了代码层面的优化策略,包括编码最佳实践、性能瓶颈的分析与优化、以及稳定性改进措施。系统层面
网络数据包解码与分析实操:WinPcap技术实战指南
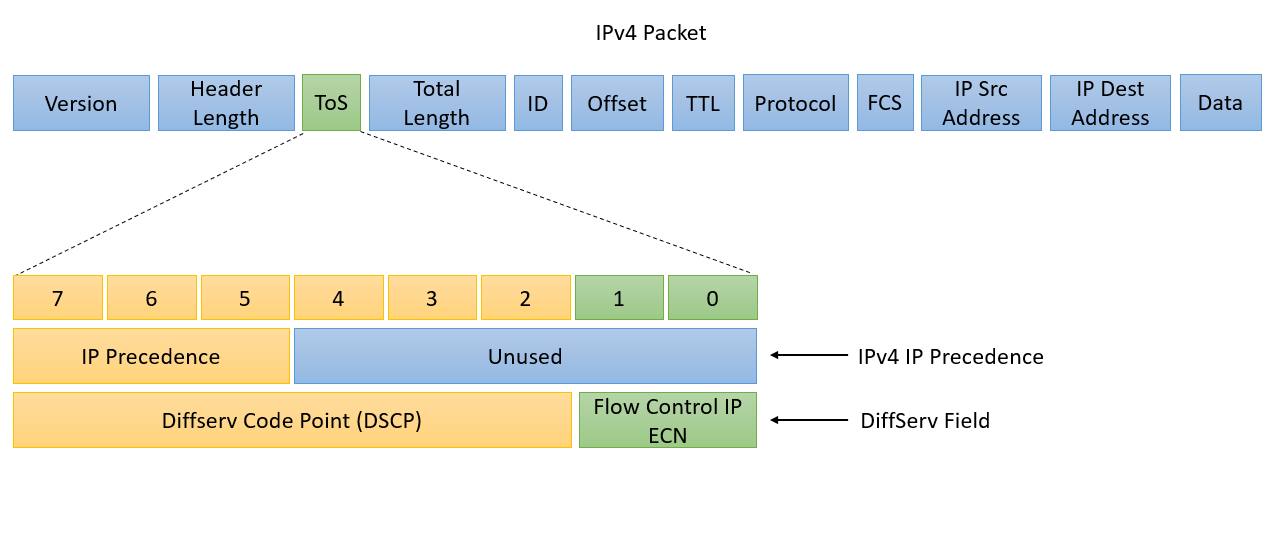
# 摘要
随着网络技术的不断进步,网络数据包的解码与分析成为网络监控、性能优化和安全保障的重要环节。本文从网络数据包解码与分析的基础知识讲起,详细介绍了WinPcap技术的核心组件和开发环境搭建方法,深入解析了数据包的结构和解码技术原理,并通过实际案例展示了数据包解码的实践过程。此外,本文探讨了网络数据分析与处理的多种技术,包括数据包过滤、流量分析,以及在网络安全中的应用,如入侵检测系统和网络
【EMMC5.0全面解析】:深度挖掘技术内幕及高效应用策略

# 摘要
EMMC5.0技术作为嵌入式存储设备的标准化接口,提供了高速、高效的数据传输性能以及高级安全和电源管理功能。本文详细介绍了EMMC5.0的技术基础,包括其物理结构、接口协议、性能特点以及电源管理策略。高级特性如安全机制、高速缓存技术和命令队列技术的分析,以及兼容性和测试方法的探讨,为读者提供了全面的EMMC5.0技术概览。最后,文章探讨了EMMC5.0在嵌入式系统中的应用以及未来的发展趋势和高效应用策略,强调了软硬
【高级故障排除技术】:深入分析DeltaV OPC复杂问题

# 摘要
本文旨在为DeltaV系统的OPC故障排除提供全面的指导和实践技巧。首先概述了故障排除的重要性,随后探讨了理论基础,包括DeltaV系统架构和OPC技术的角色、故障的分类与原因,以及故障诊断和排查的基本流程。在实践技巧章节中,详细讨论了实时数据通信、安全性和认证
手把手教学PN532模块使用:NFC技术入门指南
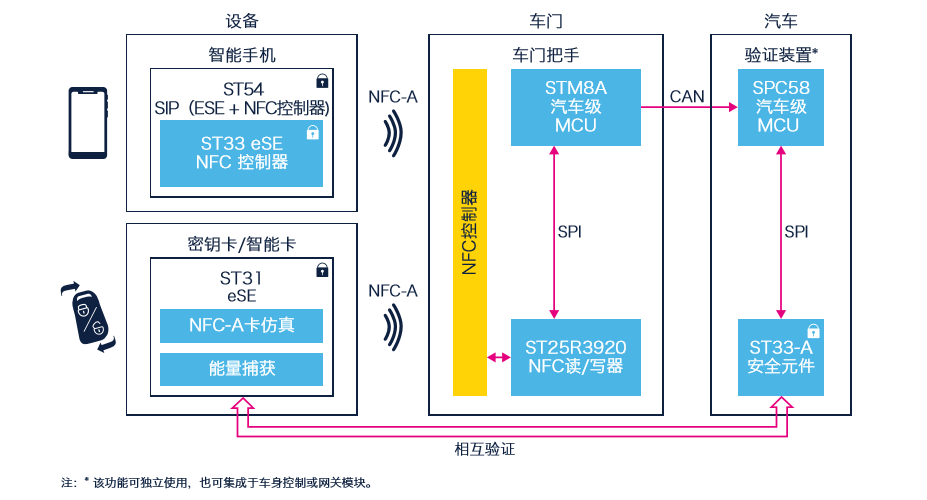
# 摘要
NFC(Near Field Communication,近场通信)技术是一项允许电子设备在短距离内进行无线通信的技术。本文首先介绍了NFC技术的起源、发展、工作原理及应用领域,并阐述了NFC与RFID(Radio-Frequency Identification,无线射频识别)技术的关系。随后,本文重点介绍了PN532模块的硬件特性、配置及读写基础,并探讨了
PNOZ继电器维护与测试:标准流程和最佳实践

# 摘要
PNOZ继电器作为工业控制系统中不可或缺的组件,其可靠性对生产安全至关重要。本文系统介绍了PNOZ继电器的基础知识、维护流程、测试方法和故障处理策略,并提供了特定应用案例分析。同时,针对未来发展趋势,本文探讨了新兴技术在PNOZ继电器中的应用前景,以及行业标准的更新和最佳实践的推广。通过对维护流程和故障处理的深入探讨,本文旨在为工程师提供实用的继电器维护与故障处
【探索JWT扩展属性】:高级JWT用法实战解析

# 摘要
本文旨在介绍JSON Web Token(JWT)的基础知识、结构组成、标准属性及其在业务中的应用。首先,我们概述了JWT的概念及其在身份验证和信息交换中的作用。接着,文章详细解析了JWT的内部结构,包括头部(Header)、载荷(Payload)和签名(Signature),并解释了标准属性如发行者(iss)、主题(sub)、受众(aud
Altium性能优化:编写高性能设计脚本的6大技巧

# 摘要
本文系统地探讨了基于Altium设计脚本的性能优化方法与实践技巧。首先介绍了Altium设计脚本的基础知识和性能优化的重要性,强调了缩短设计周期和提高系统资源利用效率的必要性。随后,详细解析了Altium设计脚本的运行机制及性能分析工具的应用。文章第三章到第四章重点讲述了编写高性能设计脚本的实践技巧,包括代码优化原则、脚
Qt布局管理技巧

# 摘要
本文深入探讨了Qt框架中的布局管理技术,从基础概念到深入应用,再到实践技巧和性能优化,系统地阐述了布局管理器的种类、特点及其适用场景。文章详细介绍了布局嵌套、合并技术,以及
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录

文章持续更新中,敬请期待~
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )