【VMD高级应用详解】:生物大分子动力学模拟分析实战指南
发布时间: 2024-12-15 06:34:08 阅读量: 3 订阅数: 1 

VMD软件教程(分子模拟)
参考资源链接:[VMD 1.8.3中文教程:从入门到高级应用](https://wenku.csdn.net/doc/84ybcs0675?spm=1055.2635.3001.10343)
# 1. VMD软件概述与基础操作
## 1.1 VMD软件介绍
VMD(Visual Molecular Dynamics)是一个开源的分子可视化和分析软件,特别在生物物理学和化学领域广泛应用。该软件可以展示生物大分子结构、模拟数据的动画及进行一系列的分析操作。
## 1.2 VMD的安装与运行
对于不同操作系统,VMD提供了详细的安装指南。以Linux系统为例,只需下载源代码并使用`make`命令进行编译安装。安装完成后,通过在终端输入`vmd`即可启动软件。
## 1.3 VMD的界面和基本操作
VMD界面被分为多个模块,如图形窗口、控制台、动画面板等。基本操作包括文件导入、视图控制、分子选择等。用户可以通过图形用户界面操作,也可以使用内置的Tcl脚本语言进行编程控制,提高操作的灵活性。
## 1.4 VMD的初学者实践指南
对于初学者来说,可以从以下几个步骤开始学习VMD:
1. 学习如何导入和渲染常见格式的分子结构文件,例如PDB文件。
2. 练习基本的视图操作,如旋转、缩放和平移。
3. 学习如何使用选择命令来高亮显示特定的分子部分或原子。
4. 尝试通过内置的帮助系统来获得更多关于特定功能或命令的信息。
以上内容是对VMD软件的一个快速概述,随后章节将深入探讨VMD在生物大分子模型构建、动力学模拟和数据分析中的应用。
# 2. 生物大分子模型的构建与可视化
生物大分子如蛋白质、核酸等构成了生命活动的基本单元。为了深入理解它们的结构与功能,研究者常借助分子模拟与可视化技术。本章将深入探讨如何使用VMD(Visual Molecular Dynamics)软件,构建并可视化生物大分子模型。
## 2.1 分子模型的导入与渲染
生物大分子模型可以来源于多种数据库,如PDB(Protein Data Bank)或自建模型。VMD软件支持多种文件格式的导入,并提供强大的渲染工具以呈现模型的细节和美感。
### 2.1.1 导入不同格式的生物分子文件
VMD支持广泛的分子模型文件格式,如PDB、PSF、PRM等。用户可以通过File菜单中的"New Molecule"选项导入文件。导入后,系统会自动识别分子类型并将其加载到VMD的主视图中。
```tcl
mol new 1XYZ.pdb type pdb
```
上述代码块展示了如何通过Tcl脚本语言导入一个名为"1XYZ.pdb"的蛋白质结构文件。这不仅展示了操作的简便性,还说明了如何通过自动化脚本批量处理多个分子文件。
### 2.1.2 设置分子模型的视觉样式和颜色方案
导入分子模型后,可以通过VMD提供的可视化工具设置分子的视觉样式和颜色方案。例如,可以改变球棍模型的半径大小,调整原子和键的颜色、渐变等属性,以适应不同的视觉分析需求。
```tcl
mol representation CPK 0.3 1.0 12
mol color Name
```
上述代码块中的第一个命令`mol representation`用于设置分子的可视化样式为CPK(Corey-Pauling-Koltun),半径大小为0.3,光照条件为1.0,球体的细分等级为12。第二个命令`mol color Name`设置根据原子类型对分子进行着色。
## 2.2 分子动力学模拟数据的可视化
分子动力学(MD)模拟是研究生物大分子动态行为的重要工具。VMD软件不仅可以导入MD模拟数据,还可以制作时间序列动画,并在三维空间中展示模拟轨迹。
### 2.2.1 时间序列动画的制作与分析
时间序列动画能够帮助研究者直观地观察到生物大分子随时间变化的过程。通过VMD的动画制作功能,可以对MD模拟轨迹文件(如XTC或DCD格式)进行操作。
```tcl
animate write psf newmol.psf
animate write dcd newmol.dcd beg 0 end 1000 waitfor all
```
上述代码块中的两个命令分别用于创建一个psf文件和一个包含从第0帧到第1000帧的轨迹动画的dcd文件。其中`waitfor all`参数确保了在生成动画之前,所有的帧已经被加载到内存中。
### 2.2.2 模拟轨迹的三维空间展示技巧
在VMD中,用户可以使用图形界面或脚本命令来展示MD模拟轨迹的三维空间特性。例如,可以利用`graph`插件来创建轨迹的空间分布图。
```tcl
package require graph
set win [graph .graph -title "Trajectory Distribution" -geometry {600 300}]
graph $win data {time x y z} \
-xLabel "Frame" -yLabel "Position" -plotcmd {plot}
```
上述代码块首先加载了`graph`包,然后创建了一个名为`.graph`的窗口,用于显示一个名为"Trajectory Distribution"的图表。图表中包含的`x`、`y`和`z`坐标是模拟轨迹的时间、位置信息。代码块还设置了图表的标题、尺寸和轴标签。
## 2.3 分子动力学模拟中的分析工具
为了深入理解分子动力学模拟数据,VMD提供了一系列的分析工具。这些工具可以用于分析生物大分子的构象变化、运动特性、相互作用等信息。
### 2.3.1 分析生物大分子构象变化
生物大分子在MD模拟过程中可能会经历构象的变化。VMD的分析工具可以帮助研究者识别这些变化并进行定量描述。
```tcl
measure minmax $mol
measure rmsd $mol
```
上述代码块中的两个命令分别用于计算分子的最小体积矩和最大体积矩以及分子构象的均方根偏差(RMSD)。这些分析结果对于理解分子的稳定性至关重要。
### 2.3.2 测定分子动力学模拟的关键参数
为了评估MD模拟的质量和可靠性,需要计算一系列关键参数,如温度、压力、能量等。VMD提供了相应的命令来快速获取这些参数。
```tcl
set temperature [measure temp $mol]
set pressure [measure pressure $mol]
set energy [measure energy $mol]
```
上述代码块展示了如何使用Tcl脚本语言在VMD中测量模拟系统的温度、压力和能量。这些参数对于模拟结果的验证和后续分析至关重要。
接下来,我们将继续探讨VMD在生物大分子模拟中的高级应用。
# 3. VMD在生物大分子模拟中的高级应用
## 3.1 分子动力学模拟的参数设置与优化
分子动力学模拟是生物大分子研究中不可或缺的工具,它可以通过模拟大分子的运动来预测其功能和行为。VMD作为一款功能强大的分子可视化和分析软件,其在参数设置与模拟优化方面的作用尤为重要。
### 3.1.1 探索最佳模拟参数的策略
模拟参数的选择直接影响模拟的精度和效率。在VMD中,用户需要根据研究目的、模拟系统和计算资源来决定参数。一个典型的工作流程包括:确定时间步长、系统温度、压力控制、能量最小化步骤、以及平衡和生产阶段的时长等。
- **时间步长**:选择一个足够小的时间步长以确保数值稳定性,通常为2飞秒。
- **温度与压力控制**:通过恒温器和恒压器来控制模拟过程中的系统温度和压力。
- **能量最小化**:在开始动力学模拟前,通常要进行能量最小化以消除分子内部的不合理的应变。
用户可利用VMD的内置功能进行模拟参数的优化,例如,可以使用VMD提供的NAMD插件,它允许用户直接在VMD中设置和运行NAMD模拟。NAMD是一个广泛使用的高性能分子动力学模拟软件。
### 3.1.2 利用VMD进行模拟系统的能量最小化
能量最小化是模拟开始前的重要步骤,目的是使分子系统达到一个能量较低的稳定状态。VMD的NAMD插件提供了方便的能量最小化操作,用户只需通过简单的图形界面就能设置最小化参数,并监控整个过程。
- 首先,用户需要加载分子模型到VMD,并确保其结构正确无误。
- 接下来,在NAMD插件中设置最小化相关的参数,例如步数、收敛条件等。
- 最后,运行最小化过程,并通过查看能量下降曲线来判断是否达到稳定。
在VMD中能量最小化的过程可以通过以下代码块实现,示例中展示了如何设置NAMD模拟最小化部分的参数:
```tcl
# NAMD configuration file snippet for minimization
minimize 1000
tolerance 1.0
```
该代码段设置了一次性的1000步最小化过程,目标能量变化的容忍度为1.0。最小化完成后,应检查分子结构和能量输出,确保达到了预期的稳定状态。
## 3.2 分子表面与通道分析
分子表面分析对理解生物大分子的功能非常重要,它可以用来识别分子的活性位点、分析分子通道和孔道特性,这些信息对于药物设计和分子识别过程尤为关键。
### 3.2.1 探索分子表面特性与识别活性位点
VMD提供了多种分析分子表面的工具,如VolMap插件,可以用来生成和可视化分子表面的密度图。VolMap能够帮助研究者识别分子表面的空腔,进而发现可能的活性位点。
- VolMap允许用户选择不同的原子类型和属性来计算表面。
- 用户可以调节VolMap的参数,例如网格大小和插值方法,以获得高质量的表面图像。
下面是一个使用VolMap分析分子表面并生成密度图的代码示例,该代码展示了如何使用Tcl脚本语言在VMD中创建一个简单的分子表面密度图:
```tcl
# VolMap configuration for generating surface density map
mol new protein.pdb
set sel [atomselect top all]
volmap density $sel -res 0.5 -minmax {{0 1} {0 1} {0 1}} -max 5.0
volmap measure $sel
```
这个脚本首先加载了一个蛋白质PDB文件,然后选择全部的原子并使用VolMap计算分子表面的密度分布,其中`-res`参数控制网格的分辨率,`-max`参数定义了密度的最大值。
### 3.2.2 分析分子通道和孔道的属性
分子通道和孔道分析有助于研究生物大分子的传输机制,这在研究膜蛋白时尤其重要。VMD的Channel Analyst插件提供了这样的功能,它不仅可以识别孔道,还可以分析孔道的属性,比如大小、形状以及位置等。
- 通道分析的第一步是创建一个蛋白质的三维体积表示。
- 使用Channel Analyst插件的特定功能来识别孔道,并提取孔道的详细信息。
下面是一个简单的示例,用于说明如何在VMD中识别和分析一个蛋白质的通道:
```tcl
# Channel Analyst configuration for detecting and analyzing molecular channels
mol load psf protein.psf pdb protein.pdb
set sel [atomselect top protein]
set p1 [measure center $sel]
set p2 [vecadd $p1 [list 0 0 20]] ;# Define the direction vector for channel search
channel analyst $sel -o $p1 -d $p2 -oresolution 0.5 -dresolution 0.5 -maxradius 10.0
```
在这个示例中,首先加载蛋白质的PSF和PDB文件,然后定义蛋白质的选择,并设定搜索通道的方向和半径参数,最后执行通道分析。结果可以被进一步可视化和分析,以获取通道的物理和化学属性。
## 3.3 VMD在跨膜蛋白模拟中的应用
跨膜蛋白是生命活动中不可或缺的一部分,它们常常嵌入在细胞膜中,执行诸如信号传导、物质运输等关键功能。VMD软件在模拟跨膜蛋白方面提供了一系列的工具和功能,使得研究人员可以更好地构建和分析跨膜蛋白模型。
### 3.3.1 构建跨膜蛋白的模拟系统
构建跨膜蛋白模拟系统的第一步是确定跨膜域。这通常可以通过生物信息学工具预测跨膜螺旋的位置。然后,使用VMD来可视化跨膜蛋白,并手动或使用内置工具调整蛋白与膜的相对位置,保证膜蛋白正确地插入到膜模型中。
- 在VMD中,可以通过加载相应的PSF和PDB文件来展示跨膜蛋白。
- 使用VMD中的分子操作工具,比如旋转和平移,调整蛋白的方向和位置。
- 利用VMD内置的构建工具,比如`membrane`插件来创建一个脂质双层,并将蛋白质嵌入其中。
### 3.3.2 分析跨膜蛋白的动态行为和功能
一旦跨膜蛋白被正确地构建并放置在模拟系统中,研究人员就需要分析其动态行为和功能。VMD中提供了多样的分析工具和脚本,可以用来研究蛋白质的构象变化、跨膜通道的运输特性和蛋白与其他分子的相互作用。
- 利用VMD的轨迹分析工具,如`measure rmsd`命令来追踪蛋白质在模拟过程中的构象变化。
- 使用`VolMap`工具分析跨膜蛋白中的孔道和通道。
- 通过分子动力学模拟,例如使用NAMD进行模拟,并在VMD中分析结果数据。
在分析跨膜蛋白的过程中,研究人员可以利用VMD的可视化能力,将不同模拟步骤的蛋白质构象叠加起来,以便观察和比较蛋白质在不同状态下的结构变化。以下是一个简单的VMD脚本示例,用于分析跨膜蛋白构象的变化:
```tcl
# VMD script snippet for analysis of transmembrane protein conformations
mol load psf protein.psf dcd trajectory.dcd
animate goto 0
set all [atomselect top all]
set initial_conformation [$all get {x y z}]
animate goto end
$all set {x y z} $initial_conformation
measure rmsd $all
```
该脚本首先加载PSF和DCD文件,代表蛋白质的初始状态。然后,通过动画命令查看到最后一个构象,最后使用measure rmsd命令比较蛋白的初始和结束构象,从而分析蛋白的动态行为。
通过VMD的高级功能,研究人员可以进行深入的生物大分子模拟分析。从模拟参数的精心设置到分子表面和通道的详细分析,再到跨膜蛋白功能的深入理解,VMD为生物大分子的研究提供了一个强大的平台。
# 4. VMD脚本语言的深入应用
VMD脚本语言,基于Tcl(Tool Command Language),为生物大分子模拟提供了强大的自动化和自定义分析能力。本章节将深入介绍VMD脚本语言的基础和进阶应用,以及如何通过编写脚本实现自动化分析和数据处理。
### 4.1 VMD脚本语言的基础与进阶
#### 4.1.1 掌握Tcl脚本语言在VMD中的应用
Tcl是一种动态编程语言,广泛应用于快速原型开发、脚本编写、GUI开发、系统管理等。在VMD中,Tcl用于编写自定义的插件、执行复杂的分析任务、自动化重复操作以及扩展VMD的功能。
**代码块示例**
```tcl
# 示例:加载一个PDB文件并显示其内容
mol new 1XYZ.pdb
```
**参数说明与逻辑分析**
- `mol new`:这个Tcl命令告诉VMD加载一个新的分子。参数`1XYZ.pdb`指定了要加载的PDB文件名。
- `1XYZ.pdb`:这是一个蛋白质数据银行(Protein Data Bank)格式的文件,包含了生物大分子的三维结构信息。
Tcl脚本的编写需要遵循VMD的命令语法,熟悉这些命令对于高级用户来说是必需的。VMD提供了详细的命令文档和在线帮助,帮助用户快速掌握所需的命令和选项。
#### 4.1.2 高级脚本编写技巧与案例分析
随着对Tcl脚本语言理解的加深,用户可以编写更复杂的脚本来实现高级功能。这包括但不限于:自动化模拟分析、数据提取和可视化等。
**高级脚本示例**
```tcl
# 示例:自动化分析蛋白质的RMSD
proc calculate_rmsd {mol} {
set all [atomselect $mol all]
set all_coords [$all get {x y z}]
$all delete
return $all_coords
}
# 假设已经加载了一个模拟轨迹
mol addfile trajectory.dcd waitfor all
# 获取初始坐标
set initial_coords [calculate_rmsd 0]
# 计算每个时间点的RMSD
for {set i 0} {$i < [molinfo 0 get numframes]} {incr i} {
mol frame $i
set current_coords [calculate_rmsd 0]
set rmsd [veclength [vecsub $initial_coords $current_coords]]
puts "Frame $i: RMSD = $rmsd"
}
```
**参数说明与逻辑分析**
- `proc calculate_rmsd {mol}`:定义一个名为`calculate_rmsd`的过程,用于计算指定分子的RMSD值。
- `set all [atomselect $mol all]`:使用`atomselect`命令选择分子中的所有原子。`$mol`是一个变量,表示分子标识符。
- `$all get {x y z}`:获取当前选定原子的坐标。
- `for`循环和`mol frame`命令用于遍历模拟轨迹中的所有帧,并计算每一帧的RMSD。
- `set rmsd [veclength [vecsub $initial_coords $current_coords]]`:计算初始坐标和当前坐标之间的距离,得到RMSD值。
- `puts "Frame $i: RMSD = $rmsd"`:打印每一帧的RMSD值。
通过编写这样的脚本,用户可以自动化处理复杂的分析任务,省去重复的手动操作,提高工作效率。
### 4.2 自动化分析与数据处理流程
#### 4.2.1 设计自动化分析脚本
自动化分析脚本是将分析流程编码化,允许用户以编程方式执行一系列任务,包括数据提取、分析计算以及结果的图形化展示。
**代码块示例**
```tcl
# 示例:自动化绘制RMSD图
# 假设已经计算了RMSD数据并存储在变量rmsd中
set rmsd_data {
{0 0.12} {10 0.14} {20 0.16} ... {100 0.15}
}
# 使用VMD内置的绘图命令
set win [mol new]
mol addrep $win
graphics $win color red
foreach {frame rmsd} $rmsd_data {
graphics $win cylinder $frame $rmsd radius 0.5
}
```
**参数说明与逻辑分析**
- `set rmsd_data {...}`:手动定义了一组RMSD数据点,每对值由帧索引和RMSD值组成。
- `foreach {frame rmsd} $rmsd_data`:使用`foreach`命令遍历RMSD数据,其中`frame`和`rmsd`分别代表每对数据中的两个值。
- `graphics $win cylinder $frame $rmsd radius 0.5`:使用`graphics`命令绘制一个圆柱体,其位置由帧索引和RMSD值决定,半径固定为0.5。
这段脚本简单演示了如何使用Tcl语言在VMD中创建一个简单的3D图示。实际应用中,RMSD数据会从模拟轨迹中自动计算并存储在数组或列表中,然后进行绘制。
#### 4.2.2 从模拟数据中提取有价值信息
从模拟数据中提取有价值的信息是生物大分子模拟的关键。这通常需要对数据进行深入分析,并将分析结果可视化。
**代码块示例**
```tcl
# 示例:分析蛋白质表面的溶剂可及性
# 假设加载了一个蛋白质分子
mol new protein.pdb
set sel [atomselect top all]
set sasa [measure sasa 2.0 [lindex [$sel get {x y z}] 0] 400 [atomselect top water]]
puts "SASA: $sasa"
```
**参数说明与逻辑分析**
- `measure sasa 2.0 [lindex [$sel get {x y z}] 0] 400 [atomselect top water]`:`measure`命令计算溶剂可及表面积(SASA)。参数`2.0`是溶剂探针半径,`[lindex [$sel get {x y z}] 0]`是蛋白质原子的坐标,`400`是模拟水分子数量,`[atomselect top water]`选择了水分子作为环境。
- `puts "SASA: $sasa"`:将计算得到的SASA值输出到控制台。
通过这样的分析,用户可以了解蛋白质表面如何暴露于溶剂,这对于理解蛋白质-溶剂相互作用非常重要。
通过本章节的介绍,VMD脚本语言在生物大分子模拟分析中的应用已经呈现得比较全面。掌握Tcl脚本不仅可以提高分析效率,还可以通过编程实现更复杂的分析任务。接下来的章节将继续探讨VMD的高级应用和与其它软件的集成。
# 5. VMD与其他模拟软件的集成应用
在生物大分子动力学模拟领域,单独的软件很难提供完整的解决方案。集成不同的模拟工具不仅可以提高工作效率,而且对于复杂问题的解决也更为有效。本章将深入探讨VMD如何与其他模拟软件,如NAMD和GROMACS进行集成,并介绍如何协同使用这些高级模拟分析工具。
## 5.1 VMD与分子动力学模拟软件的接口
VMD不仅是分子可视化领域的利器,它还能作为接口与多种分子动力学模拟软件相连接。这种集成方式可实现模拟数据的直接处理和分析,极大地提升了研究效率。
### 5.1.1 VMD与NAMD的集成
VMD与NAMD同为著名的分子模拟工具,二者之间的集成应用是生物大分子模拟领域的常用工作流之一。VMD能够加载NAMD的模拟轨迹文件,实时观察和分析模拟过程,也可以读取NAMD的输出结果进行后处理分析。
```tcl
mol new namd_output.dcd
animate goto 1000
# 上面的Tcl脚本命令在VMD中加载了NAMD的输出轨迹文件,跳转到第1000帧进行显示。
```
在集成使用时,我们要注意几个关键点。首先,确保NAMD版本和VMD版本兼容。其次,利用VMD的“extensions”菜单下的“NAMD Energy”工具进行能量分析和其它物理属性的提取。最后,可以通过编写Tcl脚本来实现自动化分析,进而获取更多深入的模拟数据。
### 5.1.2 VMD与GROMACS的数据处理
GROMACS是另一种广泛使用的分子动力学模拟软件。VMD通过读取GROMACS的XTC和TRR格式的轨迹文件来实现与GROMACS的集成。这种方式不仅能够处理大型模拟系统,还能利用VMD的可视化工具来增强分析结果的直观性。
```tcl
mol load psf protein.psf dcd gmx_output.xtc
# 该命令加载了由GROMACS产生的轨迹文件,与之前提到的NAMD轨迹文件不同,GROMACS使用XTD文件格式。
```
VMD与GROMACS集成后,用户可利用VMD内置的分析工具来评估模拟质量,比如计算均方根偏差(RMSD),对蛋白质结构的变性过程进行可视化,甚至可以进行氢键分析、溶剂可及表面积(SASA)计算等。
## 5.2 高级模拟分析工具的协同使用
集成不同模拟软件的主要优势在于可以利用各自软件的优势来处理复杂问题。下面将介绍如何利用VMD整合多来源模拟数据,并探讨VMD在跨尺度模拟中的作用。
### 5.2.1 利用VMD整合多来源模拟数据
在生物大分子动力学研究中,一个模拟项目可能会涉及多种类型的模拟,如全原子模拟、粗粒化模拟、跨膜模拟等。VMD的多功能性使其成为整合这些模拟数据的理想工具。
在实际操作中,用户可以将不同模拟的数据并行加载到VMD中,并利用VMD的分析工具来统一处理这些数据。这包括但不限于动画同步播放、参数同步修改、跨模态数据比较等。
### 5.2.2 VMD在跨尺度模拟中的角色
跨尺度模拟是分子模拟中非常重要的一个方向,它能将不同尺度的模型有效结合起来。VMD作为一个强大的可视化工具,它在跨尺度模拟中的作用不可小觑。
为了更好地展示VMD在跨尺度模拟中的应用,这里使用Mermaid流程图来描述整合多个尺度模拟数据的工作流程:
```mermaid
graph TD
A[开始] --> B[定义模拟目标]
B --> C[选择合适的模拟软件]
C --> D[进行全原子模拟]
C --> E[进行粗粒化模拟]
D --> F[使用VMD加载全原子模拟数据]
E --> G[使用VMD加载粗粒化模拟数据]
F --> H[同步多个模拟数据]
G --> H[同步多个模拟数据]
H --> I[进行跨尺度分析]
I --> J[输出可视化结果和分析报告]
J --> K[模拟结束]
```
在跨尺度模拟中,VMD不仅可以整合不同尺度的模拟数据,还可以通过其脚本语言来实现更高级的分析功能,比如制作不同尺度模拟结果的对比图、分析不同尺度下分子动态行为的差异等。
综上所述,VMD在与其他模拟软件集成应用方面发挥了巨大的作用。通过对VMD与NAMD、GROMACS等软件的集成,科研人员可以更加深入地分析和理解生物大分子的动态行为,以及它们在不同环境下的反应机制。
# 6. 生物大分子动力学模拟的案例研究
## 6.1 特定生物大分子体系的模拟分析
在生物大分子动力学模拟的实践中,选择具有特定生理功能的生物大分子进行模拟分析,有助于深入理解其在生物体内的动态行为和作用机制。本节将通过蛋白质-配体相互作用和核酸动态行为两个典型的生物大分子模拟案例,展示如何利用VMD软件进行模拟分析。
### 6.1.1 蛋白质-配体相互作用的模拟案例
在药物设计领域,理解蛋白质与配体之间的相互作用是至关重要的。以下是一个简单的模拟案例,用于展示如何利用VMD进行蛋白质-配体相互作用的分析。
1. **准备模拟系统**:首先,通过蛋白质数据银行(PDB)获取目标蛋白质的结构数据,并准备好配体分子的3D结构。
2. **模拟系统构建**:使用VMD内置的分子构建工具,将配体分子放置在蛋白质活性位点附近,并添加必要的溶剂分子和离子以平衡电荷。
3. **能量最小化**:利用VMD的模拟器进行能量最小化,以消除分子间不合理的接触。
4. **分子动力学模拟**:设置适当的力场参数和模拟条件,执行分子动力学模拟,记录蛋白质和配体的运动轨迹。
5. **分析结果**:通过VMD的分析工具,计算蛋白质与配体之间的结合能,识别关键的相互作用残基和氢键。
以下是一个VMD脚本示例,用于计算蛋白质与配体之间的距离:
```tcl
mol new protein.pdb
mol addfile ligand.pdb
set sel1 [atomselect top "protein"]
set sel2 [atomselect top "ligand"]
set distance [measure mindist $sel1 $sel2]
puts "Minimum distance between protein and ligand is: $distance"
```
在上述脚本中,`measure mindist` 命令计算了两个选择集中最近原子的距离,并输出了结果。
### 6.1.2 核酸动态行为的模拟案例
在遗传信息传递和表达过程中,核酸分子展示出复杂的动态行为。本案例将通过VMD模拟分析DNA双螺旋的热动力学行为。
1. **构建模拟系统**:导入DNA双螺旋的PDB文件,并在模拟盒内添加适当的水分子和离子。
2. **平衡体系**:通过能量最小化和预热步骤平衡体系。
3. **执行模拟**:在恒温恒压下运行MD模拟,记录DNA分子的动态变化。
4. **后处理分析**:使用VMD中的分析工具对模拟轨迹进行分析,评估DNA分子的构象变化、弯曲、扭曲等动力学特性。
为了展示热动力学分析的过程,下面的VMD脚本计算并输出DNA分子中特定原子间距离随时间的变化:
```tcl
mol new dna.pdb
animate goto 0
set sel1 [atomselect top "index 1 to 10"]
set sel2 [atomselect top "index 11 to 20"]
set file [open "distance_vs_time.dat" w]
for {set i 0} {$i < 1000} {incr i} {
animate goto [expr {$i * 0.1}]
$sel1 update
$sel2 update
set distance [measure distance $sel1 $sel2]
puts $file "$i $distance"
}
close $file
```
上述脚本通过循环模拟帧并计算两个选择集间的距离,最终将结果输出到文件`distance_vs_time.dat`中。
## 6.2 从理论到实践:模拟结果的解释与应用
模拟结果的科学解释对于理解生物分子的作用机制具有至关重要的作用,同时也为生物实验设计提供了理论依据。接下来,将探讨如何对模拟结果进行解释,并讨论其在生物实验中的潜在应用。
### 6.2.1 模拟结果的科学解释
在获得蛋白质-配体相互作用的模拟数据后,研究人员需对数据进行深入分析,以解释模拟中观察到的动态变化。例如,通过分析蛋白质与配体间的结合能,可以评估配体的亲和力;通过分析特定残基的运动模式,可以预测配体结合位点的柔性区域。
### 6.2.2 模拟数据在生物实验设计中的应用
模拟结果不仅有助于理解分子间的相互作用,而且可以指导生物实验的设计。例如,基于模拟预测的蛋白质构象变化,研究人员可以设计特定的突变实验来验证关键残基的功能。此外,模拟结果也可以用于优化药物设计中的配体结构,进而合成更加高效的药物候选分子。
本章节通过具体的案例研究,展示了如何利用VMD软件对生物大分子进行动力学模拟,并对模拟结果进行科学解释和实际应用。下一章节将更进一步,探讨如何将VMD与其它模拟软件集成,进行更为复杂和深入的分析。

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏目录

文章持续更新中,敬请期待~
最低0.47元/天 解锁专栏
买1年送1年
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【Fluent安装与配置全攻略】:第三章深入详解与最佳实践

参考资源链接:[Fluent 中文帮助文档(1-28章)完整版 精心整理](https://wenku.csdn.net/doc/6412b6cbbe7fbd1778d
【信号完整性与布线】:等长布线的原理与实践,专家级分析
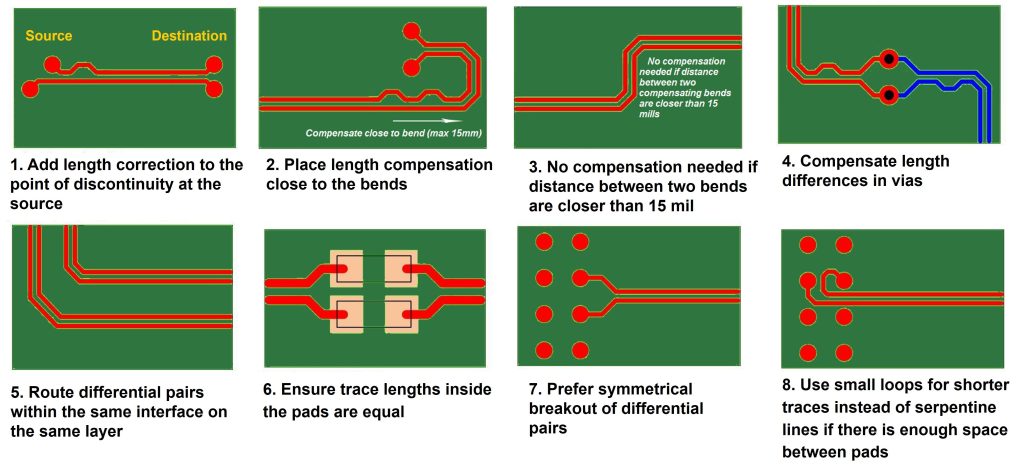
参考资源链接:[PCIe/SATA/USB布线规范:对内等长与延迟优化](https://wenku.csdn.net/doc/6412b727be7fbd1778d49479?spm=1055.2635.3001.10343)
# 1. 信号完整性与布线基础
## 1.1 信号完整性简介
在高速数
WinCC 7.2 Web发布与SCADA系统集成:实现工业自动化无缝对接

参考资源链接:[Wincc7.2Web发布操作介绍.docx](https://wenku.csdn.net/doc/6412b538be7fbd1778d425f9?spm=1055.2635.3001.10343)
# 1. WinCC 7.2 Web发布概述
随着工业4.0的推进,Web发布技术已成为连接企业与工业自动化系统的关键桥梁。WinCC 7.2作为一个工业自动化领域的强大工具,其Web发布功能为企业提供
【代码审查的艺术】:提升代码质量的有效方法

参考资源链接:[DeST学习指南:建筑模拟与操作详解](https://wenku.csdn.net/doc/1gim1dzxjt?spm=1055.2635.3001.10343)
# 1. 代码审查
【9899-202x并发编程革新】:内存模型与原子操作的全新视角
参考资源链接:[C语言标准ISO-IEC 9899-202x:编程规范与移植性指南](https://wenku.csdn.net/doc/4kmc3jauxr?spm=1055.2635.3001.10343)
# 1. 并发编程与内存模型基础
在现代计算机系统设计中,内存模型是构建高效并发程序不可或缺的基础。理解内存模型能帮助开发者编写出更加稳定、高效的并发代码。本章从基础层面探讨并发编程的基本概念,引入内存模型的概念,并简要介绍其在现代计算机系统中的重要性。
## 1.1 并发编程简介
并发编程是多线程或多进程环境下的一种编程范式。随着多核处理器的普及,合理利用并发技术已成为提升程序
【ITK-SNAP多模式应用】:不同类型图像抠图及Mask保存的策略(全面分析)
参考资源链接:[ITK-SNAP教程:图像背景去除与区域抠图实例](https://wenku.csdn.net/doc/64534cabea0840391e779498?spm=1055.2635.3001.10343)
# 1. ITK-SNAP简介及多模式图像处理基础
## 1.1 ITK-SNAP概述
ITK-SNAP是一个广泛应用于医学成像领域的开源软件,它集成了图像分割、3D注册、图像预处理等功能。其直观的用户界面和强大的算法支持,使得它在处理多模式图像时显得尤为出色。
## 1.2 多模式图像处理基础
在医学图像处理中,多模式图像指的是结合使用不同的成像技术得到的一系列图像,
【Windows 7 64位系统秘籍】:精通安装与优化SQL Server 2000的10大技巧

参考资源链接:[Windows7 64位环境下安装SQL Server 2000的步骤](https://wenku.csdn.net/doc/7du6ymw7ni?spm=1055.2635.3001.10343)
# 1
【永磁同步电机:20年经验的终极指南】:深入揭示电机性能与应用的关键

参考资源链接:[永磁同步电机电流与转速环带宽计算详解](https://wenku.csdn.net/doc/nood6mjd91?spm=1055.2635.3001.10343)
# 1. 永磁同步电机的理论基础
永磁同步电机(PMSM)以其高效率、高功率密度和优良的动态性能在现代电机技术中占据着重要地位。本章将对PMSM的基本原理和关键技术要素进行介绍,为后续章节中设计、
【Zynq-7000 SoC新手必读】:5分钟速览UG585,轻松入门Xilinx Zynq

参考资源链接:[ug585-Zynq-7000-TRM.pdf](https://wenku.csdn.net/doc/6401acf3cce7214c316edbe7?spm=1055.2635.3001.10343)
# 1. Zynq-7000 SoC概述
## Zynq-7000 SoC的架构简介
Zynq-700
【九齐单片机定时器_计数器应用】:NYIDE中高级计时技巧

参考资源链接:[NYIDE 8位单片机开发软件中文手册(V3.1):全面教程](https://wenku.csdn.net/doc/1p9i8oxa9g?spm=1055.2635.3001.10343)
# 1. 九齐单片机定时器与计数器基础
## 定时器与计数器概述
九齐单片机(如常见的9series)是微电子
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
文章持续更新中,敬请期待~
最低0.47元/天 解锁专栏
买1年送1年
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )