晶体结构建模学习路径:权威路线图助你成为行业专家
发布时间: 2024-12-26 21:39:06 阅读量: 3 订阅数: 7 

单晶硅的晶体结构建模与能带计算讲义-(1).doc
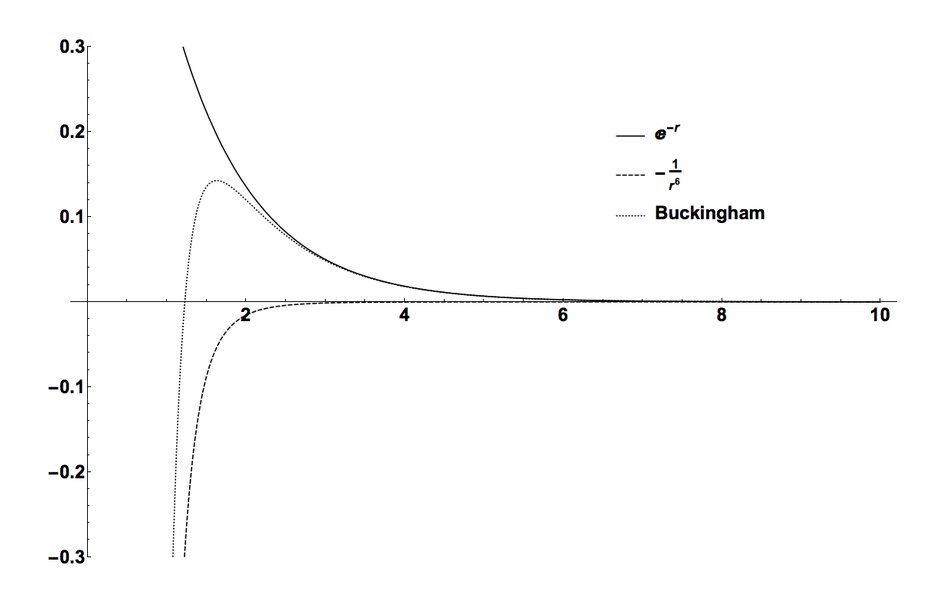
# 摘要
本文系统性地介绍了晶体结构的基础理论、建模的基本方法以及高级晶体建模技术。首先,文章回顾了晶体结构基础理论,并对晶体建模的基本方法进行了详尽的阐述,包括原子尺度建模技术和分子动力学模拟。随后,深入探讨了更先进的第一性原理计算和多尺度建模方法。在晶体结构建模实践应用章节中,详细介绍了结晶过程模拟、材料设计与优化以及计算机辅助设计软件的应用。此外,本文还分析了晶体建模在材料科学、药物设计和生物分子晶体学中的应用案例,并讨论了行业面临的挑战和未来趋势。最后,文章提供了一系列成为晶体结构建模专家的建议,涉及持续学习、研究方向选择、职业规划以及创新思维等方面。
# 关键字
晶体结构;原子建模;分子动力学;密度泛函理论;第一性原理计算;材料设计优化
参考资源链接:[CrystalMaker软件中文教程:界面与操作指南](https://wenku.csdn.net/doc/3cvgsqio7q?spm=1055.2635.3001.10343)
# 1. 晶体结构基础理论
在探究晶体结构的奥秘之前,我们首先要建立一个稳固的理论基础。晶体结构的研究不仅仅是一个数学游戏,它需要物理学和化学的紧密合作,以解释原子和分子在三维空间中的有序排列。
## 1.1 固体物质的基本概念
固体物质可以是晶体或非晶体。晶体是指原子、分子或离子按照一定的几何顺序重复排列的空间结构。而非晶体则没有这种规律性的排列。了解这些基础概念,对于掌握晶体学至关重要。
## 1.2 晶体的分类
晶体可以按照它们的对称性,晶系和空间群进行分类。一个熟知的分类方法是按照晶体的七种晶系和十四种空间群进行划分。每一种分类代表了晶体结构的一种独特方式,从而决定了其物理和化学性质。
## 1.3 晶体学的数学基础
在晶体结构的研究中,数学起到了不可或缺的作用。我们使用诸如晶格参数、布拉维格点、晶胞等概念来描述晶体结构。通过数学描述,我们可以精确地解析和预测晶体的行为和性质。
在进入更高级的建模技术之前,了解这些基础理论将为我们打下坚实的基础,让我们在后续章节中更加深入地探索晶体结构的复杂世界。
# 2. 晶体建模的基本方法
## 2.1 原子尺度的建模技术
### 2.1.1 原子排列的数学描述
在晶体建模中,原子排列的数学描述是基础,它允许科学家以一种可以计算的方式表达原子在空间中的位置和排列。原子的位置可以通过晶体学中的分数坐标来表示。分数坐标基于晶胞参数,其中每一个原子的位置可以通过其相对于晶胞边界的分数位置来描述。例如,如果晶胞的三个边长分别是 a、b 和 c,那么一个原子的位置可以通过 (x/a, y/b, z/c) 来表示,其中 x、y、z 是原子在晶胞内部的位置参数。
此外,使用矩阵和向量来描述原子在晶体中的位置也是常见的数学工具。晶体结构的每一个点阵点都可以表示为一个整数倍的矩阵乘以晶格向量,这为我们提供了一种系统化的方法来处理晶体的对称性和周期性。
### 2.1.2 晶体对称性和空间群
晶体的对称性是晶体学中极为重要的一个概念,它是理解晶体结构和物理性质的关键。对称操作包括旋转、反射、倒置以及恒等操作。空间群是一种描述晶体对称性的数学模型,它能够准确地反映出晶体结构的所有对称元素。
空间群的符号通常包括一个大写字母和一个或多个数字,例如 P2/m、Pca21、I4/mmm。大写字母P表示最简单普莱纳型,而数字代表旋转轴、螺旋轴、滑移面等的组合。在建模时,通过空间群能够极大减少计算量,因为只需要定义一个最小单元(原胞)中的原子排列,对称性会保证整个晶体的结构。
## 2.2 分子动力学模拟
### 2.2.1 势能面和分子间作用力
分子动力学模拟(MD模拟)是原子尺度模拟的核心技术之一。它依赖于对原子和分子间作用力的描述,这些作用力通常通过一个数学函数(势能函数)来表达。这个势能函数描述了分子间相互作用的能量,通常包含了键长、键角、二面角、范德华力和库仑力等项。
在进行MD模拟时,首先要确定合适的势能函数。例如,对于水分子,TIP3P、TIP4P和SPC/E是常用的几种水分子模型。每个模型都有其特定的势能参数,这些参数能够确保模拟出的水分子物理和化学性质与实际观测结果相符。
### 2.2.2 分子动力学的理论基础
MD模拟的理论基础是牛顿运动定律。通过将时间离散化为小的步长,我们可以计算出每个时间步内原子的位置和速度。在模拟的每一个时间步,根据势能函数以及牛顿第二定律 F=ma(力等于质量乘以加速度),可以求解原子的运动方程。求解过程中通常使用数值积分方法,如Verlet算法、Leapfrog算法等。
当模拟开始时,首先通过随机分布或从实验数据中获得的起始配置放置原子,并给予适当的初始速度。然后计算每个原子在力的作用下随时间的演化。模拟的总时长通常取决于所研究的物理过程,从皮秒(10^-12 秒)到微秒(10^-6 秒)甚至更长。计算结果可以用于分析晶体的热力学性质、扩散性质、相变等。
## 2.3 密度泛函理论
### 2.3.1 电子结构的基本原理
密度泛函理论(DFT)是一种计算多电子系统基态性质的强大理论工具,它在现代晶体建模中扮演了重要角色。DFT的基本思想是将复杂的多电子问题转化为电子密度的单体问题,从而显著减少了计算的复杂度。电子密度可以定义为在某个空间点位置找到电子的概率密度。
DFT的核心是Hohenberg-Kohn定理,它指出系统的基态性质可以通过电子密度唯一确定。接下来,Kohn-Sham方程将复杂的电子相互作用问题转化为一个等效的非相互作用问题。通过求解Kohn-Sham方程,可以得到体系的电子密度和基态能量。
### 2.3.2 近似方法和计算工具
DFT的实施需要近似来处理交换关联泛函,这是电子间的关联效应和交换效应的数学表达。目前已有多种近似方法被提出,例如局域密度近似(LDA)、广义梯度近似(GGA)、杂化泛函等。每种近似方法都有其适用范围和优势,在实际建模过程中,需要根据所研究的体系选择合适的近似方法。
在计算方面,DFT的实施需要依赖于专业的计算软件包,例如VASP、Gaussian、Quantum ESPRESSO等。这些软件包内嵌了DFT计算的算法,能够处理从简单分子到复杂固体材料的电子结构计算。在计算时,需要设置合理的计算参数,如截断能量、k点采样、收敛标准等,并利用超级计算机或高性能工作站来完成计算任务。通过这些高级的计算工具,研究人员可以深入理解材料的电子性质,如带隙、态密度、电荷密度分布等。
# 3. 高级晶体建模技术
在第二章中,我们已经学习了晶体建模的基本方法,本章节将更深入地探讨高级晶体建模技术,包括第一性原理计算、实验数据与模拟对比,以及多尺度建模方法,这些技术对于IT和相关行业的资深从业者来说,是探索物质世界和设计新材料的重要工具。
## 3.1 第一性原理计算
### 3.1.1 Hartree-Fock方法和Post-Hartree-Fock方法
第一性原理计算是基于量子力学的基本原理,从头计算物质的性质,不需要依赖于实验参数。Hartree-Fock(HF)方法是第一性原理计算中最早的方法之一,它假设电子在多电子系统中以独立粒子的形式运动,从而简化了多体问题。
```fortran
! 示例代码块:Hartree-Fock方法的基本步骤
program HF_Calculation
implicit none
! 初始化原子核和电子的参数
! 定义波函数和动能矩阵
! 进行自洽场迭代,求解Hartree-Fock方程
! 输出电子能量和波函数
end program HF_Calculation
```
代码块展示了HF方法的计算流程,实际计算中需要使用专业软件进行矩阵运算和积分计算。
### 3.1.2 密度泛函近似和交换关联泛函
随着计算能力的提升,第一性原理计算也发展出了更为复杂的Post-Hartree-Fock方法,其中最为广泛使用的是基于密度泛函理论(DFT)的方法。DFT方法通过电子密度来描述系统,大大简化了计算过程,而交换关联泛函的引入可以更准确地处理电子间的关联效应。
```python
from ase.dft import DFT
# 设置原子结构
atoms = ase.Atoms('H2', positions=[(0, 0, 0), (0, 0, 0.74)])
# 创建DFT对象并选择交换关联泛函
dft = DFT(atoms)
dft.set(xc='pbe') # 使用PBE泛函
# 运行计算并输出结果
dft.calculate()
print('Total energy:', dft.get_potential_energy())
```
Python代码块利用ASE库进行DFT计算,展示了如何设置交换关联泛函并计算分子的总能量。
## 3.2 实验数据与模拟对比
### 3.2.1 X射线衍射和电子显微镜技术
实验数据在晶体建模中发挥着关键作用。X射线衍射(XRD)是一种确定材料晶体结构的常用技术,通过分析衍射模式可以得到晶体的晶格参数和对称性信息。而电子显微镜(TEM)技术能够在原子级别上提供直接的图像信息。
```mermaid
graph TD
A[X射线衍射] -->|获取数据| B[晶体结构分析]
A -->|校正| C[样品位置]
A -->|制备| D[样品预处理]
E[电子显微镜技术] -->|获取数据| F[图像分析]
```

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏提供晶体结构建模分析软件的全面指南,从入门到精通,涵盖实用技巧、权威指南、实例教程、软件应用、界面自定义、数据管理、故障排除、可视化技术、报告撰写、学习路径、跨学科应用、误差分析、定制开发、生物医药应用、数据可视化工具比较、教学法、性能优化、兼容性和更新等各个方面。专栏汇集了行业专家的见解和实践经验,旨在帮助读者掌握晶体结构建模软件,提升建模能力,并深入了解晶体结构建模在材料科学、生物医药等领域的应用和挑战。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
S32K144开发全攻略:零基础到精通的10大秘籍
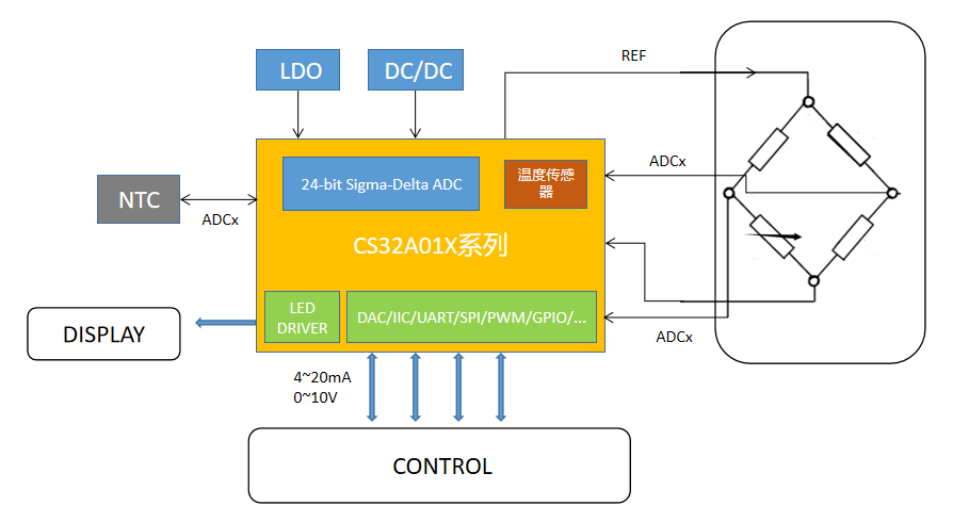
# 摘要
本文详细介绍并指导了S32K144开发板的配置、编程和优化过程,涵盖了从基础设置到高级功能实现的各个方面。文章首先介绍了开发板的基本信息和设置,然后着重于开发环境的搭建,包括CodeWarrior IDE和S32 Design Studio的配置,以及基本调试技术的掌握。在基础编程指南中,介绍了S32K144的硬件架构,演示了如何编写裸机程序和管理中断。深
【电子元器件全方位精通指南】:初级入门到专家进阶全攻略

# 摘要
电子元器件作为电子系统的基本组成单元,对电子设备的性能和稳定性起着至关重要的作用。本文从基础知识出发,对电子元器件进行了详细的分类,并深入探讨了被动元件、主动元件、机电元件和传感器的功能与应用。同时,本文提供了元器件选择与应用的技巧,以及如何在电路设计中进行有效利用。此外,文章还涵盖了电子元器件测试和故障诊断的常用技术和高级方法,以确保电子设备的可靠运行。最后,文章展
LSU4.9-BOSCH氧传感器故障速查:10个案例与高效解决法

# 摘要
氧传感器是汽车尾气排放控制系统的关键组成部分,其正常工作对于确保汽车排放符合环境标准至关重要。本文首先介绍了氧传感器的工作原理及其在汽车排放系统中的重要性。接着,详细阐述了LSU4.9-BOSCH氧传感器的故障诊断基础,包括故障诊断流程、常见故障类型及其成因、以及相应的检测工具与方法。通过10个经典案例的分析,本文提供了故障诊断的实战技巧,并分享了问题的解
机械性能测试新境界:SMTC电连接器技术深度剖析及实践应用

# 摘要
SMTC电连接器作为通信和电子系统的关键组成部分,其技术的先进性和可靠性直接关系到整体系统性能。本文首先概述了电连接器的基本概念和理论基础,详细阐述了其工作原理和性能指标,特别是电流传输机制、接触电阻及信号完整性对电连接器性能的影响。接着,本文着重介绍了SMTC电连接器的技术创新实践,包括模块化设计、高密度互连技术、高性能材料的应用,以及制造工艺的革新。此外,文中还探讨了SMTC电连接器在实验室环境和实际应用中的测试方法
【Tomcat架构揭秘】:10个技巧助你深入解读源码
# 摘要
本文对Apache Tomcat服务器的架构和性能优化技巧进行了深入探讨。首先解析了Tomcat的核心组件,包括类加载机制和连接器设计,并详细分析了其生命周期管理。接着,文章探讨了性能调优的实践方法,涉及线程模型、连接器配置以及应用部署与资源管理。文章的第四章对Tomcat的安全机制进行了探秘,包括认证与授权机制、安全漏洞分析与防范、以及SSL/TLS配置与优化。第五章讨论了如何通过插件机制与深度定制来扩展和个性化Tomcat的行为。最后,第六章通过多个实践案例分析,展示了多节点集群部署、高可用性部署策略以及从源码到生产环境的Tomcat部署技巧。本文旨在为读者提供全面的Tomcat
gprMax3.0参数优化实战:用遗传算法优化模型参数的策略

# 摘要
本文首先介绍了gprMax3.0模型和遗传算法的基本概念,然后重点探讨了遗传算法在参数优化中的理论基础,包括算法的起源、运作机制、组件与流程以及优化过程中的优势与挑战。随后,文章通过gprMax3.0模型参数优化实践,展示了遗传算法的具体应用步骤,包括问题定义、建模、编码、适应度评估以及选择、交叉和变异操作。此外,本文还提出了一
【逆变器滤波电感材料优选】:关键材料对性能的影响
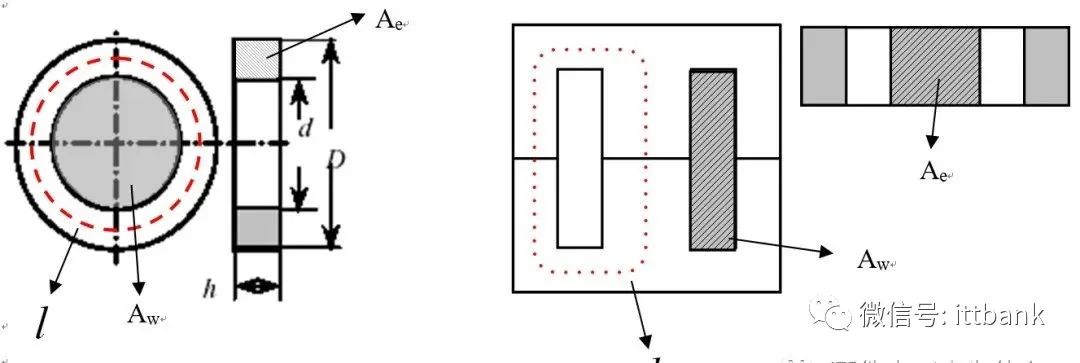
# 摘要
逆变器滤波电感作为电力电子系统中的关键组件,对改善功率质量、降低电磁干扰至关重要。本文详细介绍了逆变器滤波电感的基本概念、作用及其设计过程中的考量标准,探讨了电感材料的基础理论、性能参数、成本、可持续性和可靠性等多个维度。通过对不同电感材料的优选标准进行分析,以及实验验证和应用案例的研究,本文提出了逆变器滤波电感设计的
AI导论与实践:如何通过洗衣机实验深入理解模糊推理?

# 摘要
随着人工智能技术的快速发展,AI模糊推理技术在理论和实践领域均取得显著进展。本文从AI导论与实践的理论基础出发,重点探讨了模糊逻辑的基本原理,包括模糊集合与隶
内容安全大师:FreeCMS用户权限管理的最佳实践

# 摘要
随着信息系统的日益复杂和安全要求的不断提升,用户权限管理已成为保障系统安全和提升管理效率的关键环节。本文首先概述了用户权限管理的重要性和基础理论,然后详细介绍了FreeCMS权限管理系统的架构、身份验证机制以及角色和权限分配模型。通过实战指南,本文深入讨论了用户和角色的创建与管理、权限的分配与审核、系统安全策略及审计日志的应用。在复杂场景下的用户权限管理章节中,本文探讨了多组织结构下的权限管理策略、高级权
【企业级应用最佳实践】:如何稳定读取Word文档,避免Apache POI空指针异常
# 摘要
Apache POI是处理Microsoft Office文档的一个流行的Java库,本文详细介绍了Apache POI的基本概念、异常处理机制、高效文档读取策略以及企业级应用中的安全性和兼容性问题。通过对异常类型的深入分析以及编程策略的探讨,本文提供了实用的错误预防和调试技巧。在文档处理方面,本文不仅阐述了结构解析和高效处理方法,还提供了创建稳定文档读取应用的实例演练。最
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )