焓、熵及自由能的精准评估:Gaussian 16 B.01热力学性质计算
发布时间: 2024-12-15 23:34:08 阅读量: 65 订阅数: 28 

参考资源链接:[Gaussian 16 B.01 用户指南:量子化学计算详解](https://wenku.csdn.net/doc/6412b761be7fbd1778d4a187?spm=1055.2635.3001.10343)
# 1. Gaussian 16 B.01软件概述
## 简介
Gaussian 16 B.01是化学研究领域广泛使用的量子化学计算软件包,由Gaussian公司开发。它提供了丰富的方法来模拟分子的电子结构和动力学行为,广泛应用于有机化学、无机化学、材料科学和生物学等领域。
## 主要特点
Gaussian 16 B.01包含了多种计算方法,如Hartree-Fock、密度泛函理论(DFT)、Møller-Plesset微扰理论等。软件支持多种基组和优化算法,能够进行分子结构优化、频率分析、反应路径计算和多种谱学模拟等。
## 功能与应用
该软件不仅支持单点能量计算,还可进行热力学性质的计算,例如焓、熵、自由能和反应热等。它在研究化学反应、分子间相互作用、材料性质等方面有着重要的应用。
Gaussian 16 B.01软件是进行分子模拟和热力学性质计算的强大工具,为化学和相关科学领域的研究提供了丰富的功能和高度的灵活性。在后续章节中,我们将深入探讨其热力学计算的功能与应用。
# 2. 热力学基础与计算理论
### 焓、熵及自由能的热力学定义
在热力学中,焓、熵和自由能是描述系统能量状态和趋向平衡的基本概念。理解这些概念对于分析和计算化学和物理过程至关重要。
#### 焓的概念及其在热力学中的角色
焓是衡量系统内能和外部压力能总和的热力学量,定义为内能U加上压力P和体积V的乘积(H = U + PV)。在恒压过程中,焓变(ΔH)等于系统吸收或释放的热量。以下是焓的数学表达式:
```math
ΔH = ΔU + Δ(PV)
```
在化学反应中,焓变可以表示为反应热,这与反应在标准状态下的放热或吸热性质直接相关。
#### 熵与系统无序度的关系
熵是衡量系统无序度的量,是系统状态的函数,表示为S。熵的增加表示系统无序度的增加,符合热力学第二定律。对于一个自发过程,系统的总熵变化(ΔS总)是系统熵变和周围环境熵变之和,即 ΔS总 = ΔS系统 + ΔS环境。
熵可以使用以下方程来计算:
```math
ΔS = ∫(dq/T)
```
其中dq表示微小热交换,T是系统温度。
#### 自由能的热力学意义与平衡条件
自由能,特别是吉布斯自由能(G)和亥姆霍兹自由能(A),是用来预测系统能否自发进行某过程的热力学势。在恒温恒压下,吉布斯自由能的减小(ΔG < 0)表明过程是自发的,对于可逆过程,自由能的变化等于系统可用来做功的最大非体积功。
吉布斯自由能变化的数学描述如下:
```math
ΔG = ΔH - TΔS
```
在平衡状态下,自由能变化为零(ΔG = 0)。
### 热力学性质的计算方法
#### 统计力学基础与分子模拟
统计力学提供了从微观粒子行为到宏观物质性质的桥梁。它使用概率论和统计方法来确定宏观物理性质与微观状态的关系。分子模拟,如蒙特卡洛模拟和分子动力学模拟,是应用统计力学原理来模拟分子系统行为的有效工具。
#### 热力学性质的实验测定与理论计算对比
热力学性质的理论计算与实验测定之间需要对比验证。实验测定依赖于精确的仪器和科学的实验设计,而理论计算则依赖于正确的物理模型和适当的数学近似。
理论计算可提供对实验难以测量的复杂系统的见解,同时实验数据可以校正和优化计算模型。
#### 热力学数据库及应用实例
热力学数据库是包含大量物质热力学性质的数据集合,它们为科学研究和工程应用提供了宝贵的信息。在实际应用中,可以利用这些数据库进行物质的热力学性质预测,如相平衡、化学反应平衡常数等。
在某些专业领域,例如材料科学和化学工程,数据库的准确性和完整性是进行有效热力学计算的关键。
在本节中,我们已经详细介绍了热力学基础概念和计算方法。下一节,我们将深入探讨Gaussian 16 B.01软件中的热力学计算功能,并通过实例分析展示其在计算焓、熵和自由能方面的应用。
# 3. Gaussian 16 B.01中的热力学计算功能
## 3.1 Gaussian 16 B.01软件热力学模块介绍
### 3.1.1 热力学性质计算的软件界面
Gaussian 16 B.01是一款功能强大的量子化学计算软件,它提供了丰富的模块来进行各种化学和物理性质的计算,其中热力学性质的计算是其核心功能之一。软件界面的设计旨在为用户提供直观、易用的操作体验,同时确保科学计算的精确性与高效性。
软件的主要界面由以下几个部分组成:
- **分子构建器**:允许用户通过图形界面构建分子模型,也可以输入分子坐标以创建复杂的化学结构。
- **计算方法选择**:提供多种量子化学计算方法,如Hartree-Fock、DFT、MP2等,供用户选择,以适应不同计算需求。
- **热力学参数设置**:用户可以设置温度、压力等参数,以模拟实际实验条件下的热力学性质。
- **输出控制**:允许用户指定所需的输出内容,如热力学量、频率、振动能级等。
界面设计简洁且功能性强大,使得即使是初学者也能够快速上手。所有计算的输入文件都可以通过文本编辑器进行调整,高级用户可以利用这一特性进行更细致的设置。
### 3.1.2 热力学计算相关的输入输出说明
在Gaussian 16 B.01软件中,进行热力学计算的输入文件需要包含特定的指令集。例如,计算标准摩尔熵(S°)、标准摩尔焓(H°)、标准摩尔吉布斯自由能(G°)等热力学量时,需要使用如下指令:
```gjf
#p freq B3LYP/6-31G(d) nosymm
Title Card Required
```
这里,`#p`指令后跟的是计算方法和基组的选择,`freq`代表进行频率分析来获取热力学数据,`B3LYP/6-31G(d)`则是选定的计算方法和基组。`nosymm`表示不考虑分子对称性,通常用于更准确的频率计算。
输出文件通常非常详细,包含了热力学量的计算结果。热力学性质的输出通常位于频率计算部分之后,一般包括零点能、内能、焓、自由能等,有时也会包括温度和压力校正后的热力学数据。
### 3.2 焓、熵及自由能的计算实例分析
#### 3.2.1 不同分子模型的热力学计算策略
在实际应用中,选择合适的分子模型对于热力学计算至关重要。由于分子模型的不同,计算得到的热力学量也会有显著差异。对于小型分子,可以使用较为精确的从头算方法(如CCSD(T))和较大基组(如aug-cc-pVTZ)。对于大型分子系统,可能需要使用密度泛函理论(DFT)方法,并选用适当的混合泛函和简化基组以节省计算资源。
#### 3.2.2 实际案例的计算步骤与结果解析
以一个具体的有机反应为例,我们需要计算反应物、中间体和产物的焓、熵和自由能。以下

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
《Gaussian 16 B.01 用户参考》专栏为用户提供了全面的指南,介绍了 Gaussian 16 B.01 计算化学软件的最新特性和功能。从新特性解析到优化算法的平衡艺术,再到材料科学计算模型指南,该专栏涵盖了各种主题。此外,它还提供了安装和配置秘籍,性能测试和调优技巧,以及并行计算的实战策略。该专栏还探讨了量子化学基础、药物设计、用户社区经验分享、资源限制下的计算精度调整、数据展示和分析技巧、化学反应路径分析、污染物反应机制研究、大分子结构和功能研究、波函数和电子密度解读、热力学性质计算、光谱模拟和分析以及核磁共振谱图的理论预测。通过深入的分析和实用建议,该专栏旨在帮助用户充分利用 Gaussian 16 B.01 的强大功能,从而提升计算化学效率,并推进科学研究和创新。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【零基础到精通】:3D渲染技术速成指南,掌握关键技巧

# 摘要
本文系统地介绍了3D渲染技术,从理论基础到实际应用进行了全面阐述。首先介绍了3D渲染的基本概念、光线追踪与光栅化的原理、材质与纹理贴图的应用,以及照明与阴影技术。接着,文章深入探讨了当前流行的3D渲染软件和工具,包括软件功能和渲染引擎的选择。实践案例分析章节通过具体实例展示了产品、角色与动画以及虚拟现实和3D打印的渲染技巧。最后,文章聚焦于渲染速度提升方法、高级渲
压力感应器校准精度提升:5步揭秘高级技术
# 摘要
提升压力感应器校准精度对于确保测量准确性具有重要意义,特别是在医疗和工业制造领域。本文首先介绍了压力感应器的工作原理及其校准的基础知识,然后探讨了提高校准精度的实践技巧,包括精确度校准方法和数据分析处理技术。文章还探讨了高级技术,如自动化校准和校准软件的应用,以及误差补偿策略的优化。通过对典型行业应用案例的分析,本文最后提出了校准技术的创新趋势,指出了新兴技术在校准领域的潜在应用和未来发展方向。本文旨在为专业技术人员提供系统性的理论指导和实践经验,以提升压力感应器的校准精度和可靠性。
# 关键字
压力感应器;校准精度;自动化校准;数据分析;误差补偿;校准技术
参考资源链接:[鑫精
【24小时精通TI-LMK04832.pdf】:揭秘技术手册背后的技术细节,快速掌握关键信息
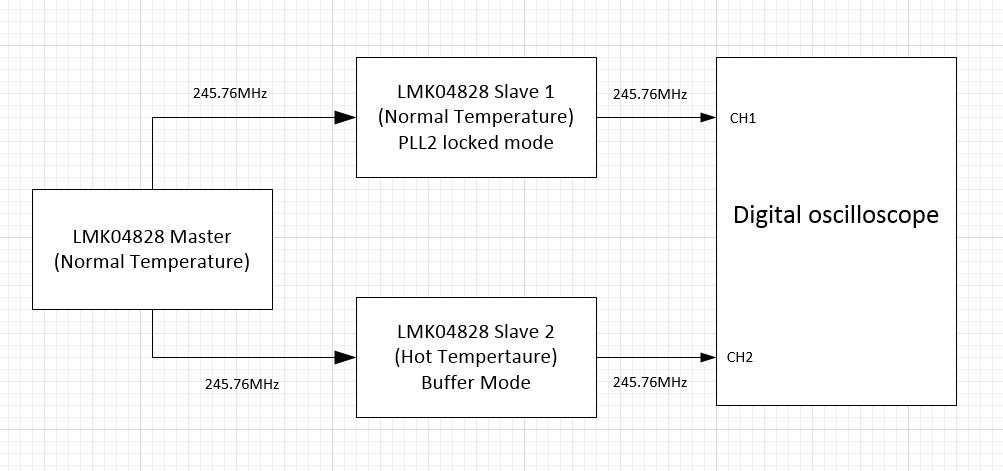
# 摘要
LMK04832是高性能的时钟发生器与分配设备,本文全面介绍其技术手册、工作原理、性能参数、应用电路设计、编程与配置,以及故障排除与维护。本手册首先为读者提供了关于LMK04832的概览,接着详细分析了其内部架构和关键性能参数,阐述了信号路径和时钟分配机制,并指
STM32电源问题诊断:系统稳定性的关键策略

# 摘要
STM32系统作为广泛应用于嵌入式领域的一个重要平台,其电源稳定性对整个系统的性能和可靠性至关重要。本文系统地分析了电源问题对STM32系统稳定性的影响
深入揭秘VB.NET全局钩子:从原理到高效应用的全攻略
# 摘要
全局钩子在软件开发中常用于监控和响应系统级事件,例如键盘输入或鼠标活动。本文首先概述了VB.NET中的全局钩子,随后深入探讨了其内部工作机制,包括Windows消息系统原理和钩子的分类及其作用。文章详细介绍了在VB.NET环境下设置和实现全局钩子的具体步骤,并通过键盘和鼠标钩子的使用案例,展示了全局钩子的实际应用。进一步,本文探讨了全局钩子在多线程环境下的交互和性能优化策略,以及安全性考量。最后,文章提供了
前端性能优化实战秘籍:10个策略让你的页面飞起来

# 摘要
随着互联网技术的快速发展,前端性能优化成为提升用户体验的关键因素。本文对前端性能优化进行了全面的概述,深入探讨了页面渲染优化技术,包括关键渲染路径、代码分割与懒加载,以及CSS优化。在资源加载与管理方面,文章分析了资源压缩与合并、异步加载及CDN加速的有效策略。进一步地,本文还讨论了交互与动画性能提升的方法,如GPU加速、动画优化技巧及交互性能调优。此外,文章还介绍了前端监控与分析工
CMW500信令测试故障排除:20个常见问题与应对策略

# 摘要
本文
CPCI标准2.0中文版数据隐私保护指南

# 摘要
本文全面介绍了CPCI标准2.0在数据隐私保护方面的应用和实践。首先概述了CPCI标准2.0的基本内容,并详细讨论了数据隐私保护的基础理论,包括其定义、重要性以及与数据保护原则的关系。随后,文章对比了CPCI标准2.0与国际数据隐私保护标准,如GDPR,并探讨了其具体要求与实践,特别是在数据主体权利保护、数据处理活动合规性及跨境数据传输规则方面。此外,本文着重阐述了CPCI标准2.0在实施过程中所依赖的技术保障措施,如数据加密、匿名
【TOAS流程优化】:OSA测试流程详解与操作步骤优化建议

# 摘要
本文针对TOAS流程的全貌进行了深入探讨,涵盖了OSA测试流程的理论与实践操作。通过对测试流程中的关键活动、要素以及测试前后的重要步骤进行分析,本文揭示了TOAS流程中常见的问题与挑战,并提供了优化建议和理论支撑。具体操作步骤包括流程映射与诊断、重构与标准化,以及监控与持续改进。文章通过案例分享,展示了TOAS流程优化的成功与失败经验,旨在为相关流程管理和优化提供
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )