BLAST算法详解:生物信息学中的序列比对工具
需积分: 37 34 浏览量
更新于2024-08-01
1
收藏 1.08MB PDF 举报
"这篇文章主要介绍了BLAST算法,一种在生物信息学中广泛使用的序列比对工具,用于在大规模数据库中快速查找与特定序列相似的序列。BLAST算法由Altschul等人在1990年提出,其设计目标是高效地识别具有局部相似性的序列。"
BLAST算法的核心思想是基于成对局部序列比对,它不是寻找全局最优的比对,而是快速找到高得分的局部匹配。这种策略大大减少了计算时间,使得在大型基因或蛋白质序列库中进行搜索成为可能。
在序列比对中,主要目的是揭示序列间的相似性和潜在的生物学意义。通过比对,我们可以推测序列间的共同区域,这些区域可能对应于相同的生物学功能或者暗示它们共享共同的祖先。成对序列比对用于分析两条序列之间的关系,而多序列比对则涉及多条序列,旨在发现所有序列中的保守区域,这对于理解蛋白质家族的结构和功能至关重要。
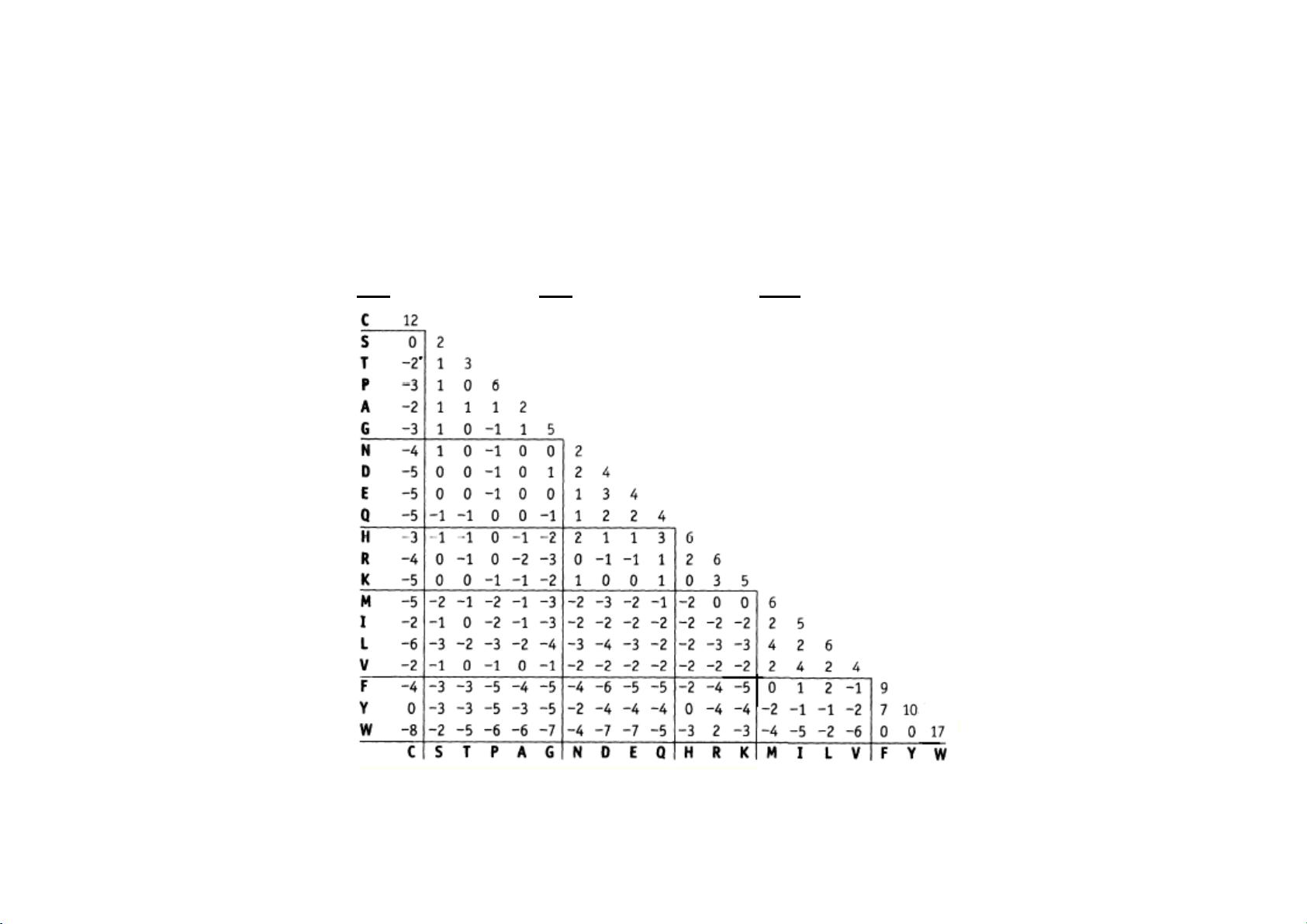
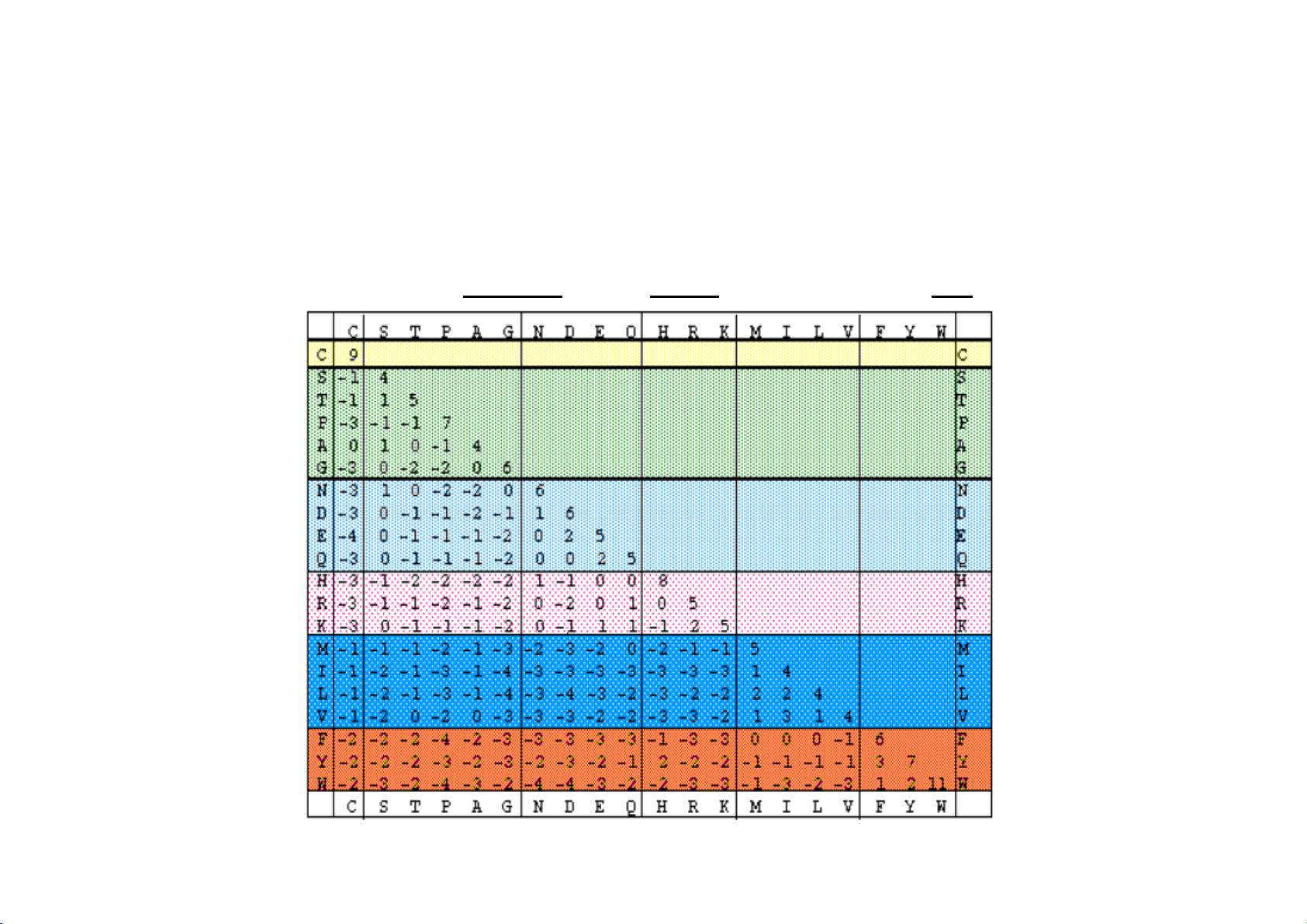
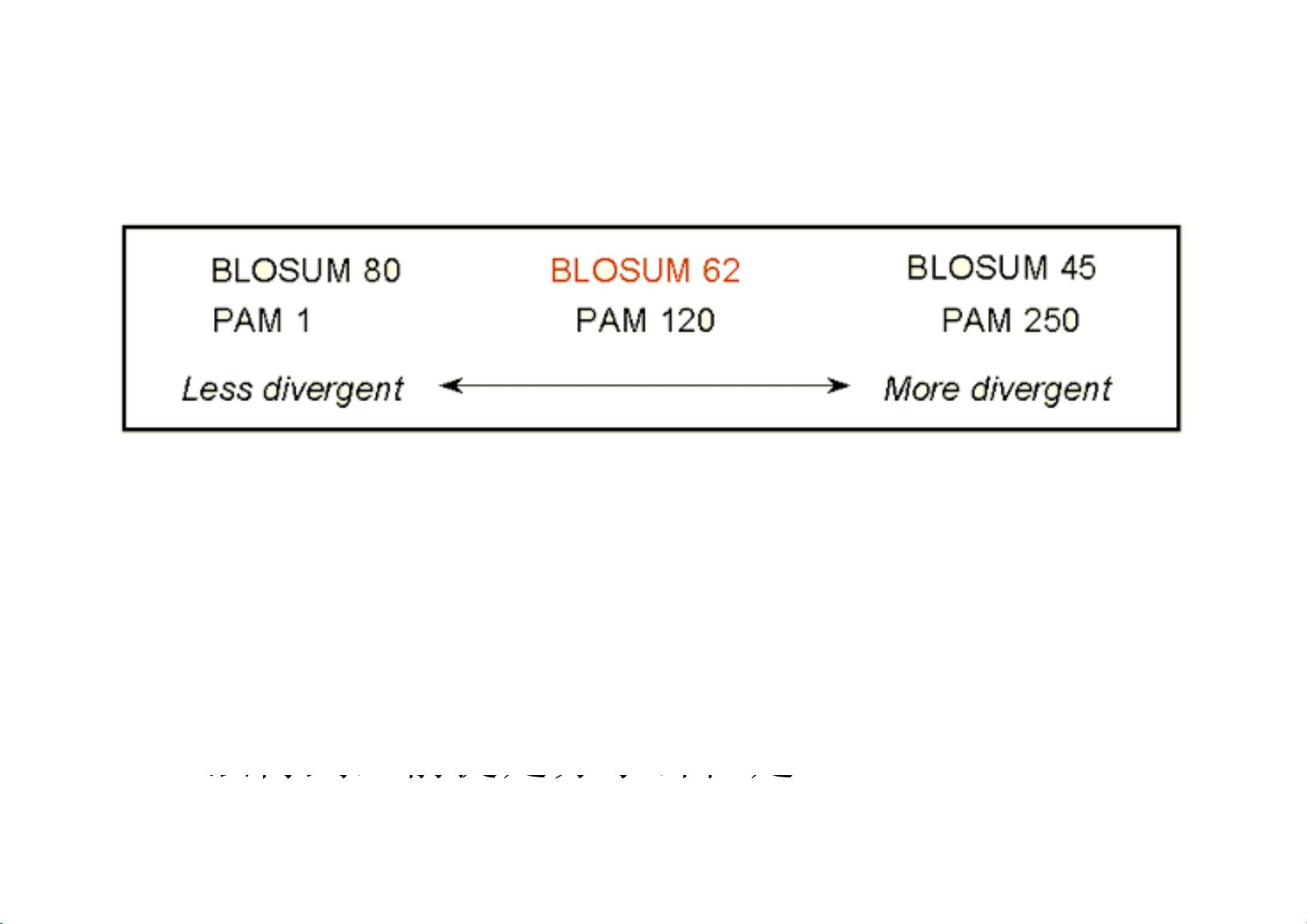
在BLAST中,比对的质量通常由一个打分函数来衡量,该函数包括打分矩阵和空位罚分两部分。打分矩阵如PAM或BLOSUM系列,用于量化不同氨基酸或碱基替换的相似性或差异。空位罚分则是为了考虑插入或删除操作的成本,因为这些操作通常意味着序列间的不匹配。在BLAST算法中,选择合适的打分系统和空位罚分策略对于准确识别相关序列至关重要。
除了基本的比对,BLAST还提供了多种变体,如blastn用于DNA-DNA比对,blastp用于蛋白质-蛋白质比对,blastx用于DNA-蛋白质翻译比对,以及tblastn和tblastx用于蛋白质到基因组的比对。这些工具各有侧重,满足了不同生物信息学研究的需求。
BLAST算法是生物信息学中一个极其重要的工具,它极大地推动了对基因和蛋白质功能的理解,尤其是在基因家族、蛋白质结构域以及进化关系的研究中。通过对大量序列进行快速比对,科学家们能够揭示生物体间的遗传关联,从而推进医学、农业和基础生物学等多个领域的研究。
2024-03-27 上传
2024-03-27 上传
2023-09-14 上传
2024-03-27 上传
2023-06-08 上传
2023-06-08 上传

passionthean
- 粉丝: 1
- 资源: 10
 我的内容管理
展开
我的内容管理
展开







最新资源
- csharpjkmemoty,c#简单mssql线程池+异步socket服务端完整源码,c#
- subclass-dance-party
- ExiFlow-开源
- Pre-2020 Google Icons-crx插件
- recipe-book:格雷格和艾莉的食谱书(v4)
- weekly_u3etas
- nCode,c#教材订购系统源码,c#
- chatterbox-client
- Wikiquote (ES)-crx插件
- 实时股票查看器:绘制和分析来自彭博或雅虎的实时市场数据。-matlab开发
- 物资管理系统项目源码.zip
- EqualitySpad.t9qmko61wz.gaF8I5O
- React横幅制作者
- I-Need-a-Hero
- main-form,c#如何将源码生成dll,c#
- investment-app:决定投资计划之前要问的问题
