癌症基因组互斥性检测:计算方法解析与肿瘤易感性揭示
34 浏览量
更新于2024-08-26
收藏 1.25MB PDF 举报
本文主要探讨了在癌症基因组学研究中识别基因互斥性的重要性和计算方法。随着癌症基因组的系统测序技术的发展,科学家们发现癌症患者往往携带有多种遗传变异的组合,这些组合模式揭示了肿瘤的复杂性。在众多癌症进程中,互斥性现象尤为显著,即一组基因的行为似乎互相排斥,可能意味着它们在疾病发展过程中扮演着互补的功能或者存在协同抑制效应。
互斥性被广泛认为反映了肿瘤细胞的进化策略,它可能揭示了某些特定基因组合下的肿瘤易感性或治疗弱点。为了深入理解这种现象,研究人员提出了多种计算方法,这些方法旨在从基因组数据中挖掘遗传交互作用,并通过整合基因网络和临床表型来揭示潜在的肿瘤特征。这些算法包括但不限于统计分析、聚类分析、网络分析以及机器学习模型,如逻辑回归、随机森林和深度学习等。
作者以系统性的视角介绍了互斥性的基本假设,例如基因互斥性可能与关键癌症通路的失调有关,或者与肿瘤抑制机制的失效相联系。文章详细阐述了识别互斥模式的策略,例如寻找在特定条件下仅有一个基因活性的实例,或者检测基因表达水平的显著负相关性。此外,作者还通过对模拟数据集的比较,评估了不同算法在性能上的优劣,以确保结果的可靠性和有效性。
然而,值得注意的是,互斥性的研究并非没有挑战。不同的计算方法可能会受到混杂因素的影响,如样本大小、数据质量、基因调控复杂性等。因此,研究人员在应用这些方法时,需要考虑如何控制和调整这些因素,以减少误差和提高解释的准确性。
最后,本文的作者——来自哈尔滨医科大学生物信息科学技术学院的研究者们,强调了互斥性研究在理解致癌机制和发现新的治疗靶点方面的潜力。他们通过通信作者的联系方式分享了他们的研究成果和进一步合作的可能性,这对于癌症研究领域来说是一项重要的贡献。
这篇文章不仅提供了关于癌症基因组互斥性现象的深入解析,还为同行提供了实用的计算工具和技术,推动了癌症生物学和精准医学的进步。
samples are covered by more than one module members (called
low impurity). Such properties can be used to infer combin-
ational scores to qualify mutual exclusivity. However, searching
for candidate mutually exclusive modules is a NP-hard problem
[44, 47, 48]. Hundreds, even thousands, of somatic alterations are
detected in certain cancer types, and their possible combinations
become enormous. There are as many as 10
8
–10
11
for combin-
ations of four genes, making exact enumeration impossible.
A feasible resolution to this dilemma is utility of heuristic algo-
rithms, such as the greedy algorithm, the Markov chain Monte
Carlo (MCMC) method and the simulated annealing algorithm
[43, 44].
Vandin et al. [44] developed Dendrix that adopts two search-
ing strategies, including the greedy algorithm and the MCMC
method, to identify mutually exclusive modules. In brief,
starting from seed genes, the greedy algorithm iteratively adds
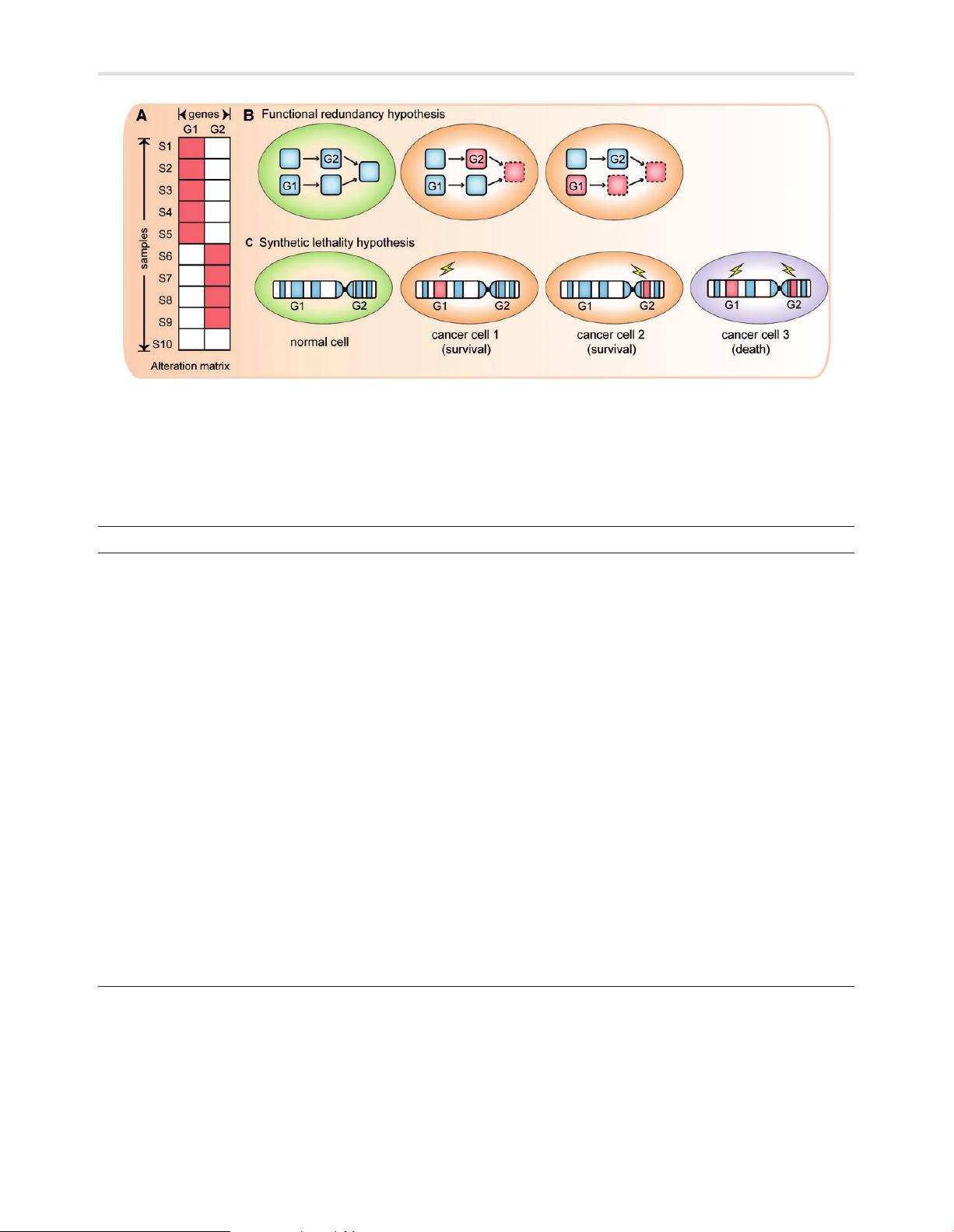
Figure 1. The common hypotheses of mutual exclusivity. (A) Mutual exclusivity between G1 and G2. (B) The functional redundancy hypothesis. Suppose G1 and G2 lie
in the same pathway, and they share the same downstream effect. Squares indicate molecular members in the pathway. While solid squares in red exhibit genetic
alterations, solid squares in blue exhibit normal activity. Dotted squares in red indicate dysfunction molecules in this pathway, which are induced by the genetic
alteration of G1 or G2. (C) The synthetic lethality hypothesis, i.e. the co-occurrence of G1 and G2 is detrimental to cancer cells. Red part shows genetic alteration.
Circles in green indicate normal cells, circles in orange indicate living cancer cells and circles in purple indicate death of cancer cells.
Table 1. Examples of mutual exclusivity with biological significance
Gene 1 Gene 2 Cancer type Phenotype Pubmed ID
BRCA1 CCNE1 Ovary cancer Synthetic lethality 24218601
KRAS TP53 Lung cancer Synthetic lethality 19075675
ATM TP53 Colorectal adenocarcinoma Synthetic lethality 22660439
BRCA2 TP53 HeLa cells; breast cancer Synthetic lethality 17000754
BRCA1 PARP1 Breast cancer Synthetic lethality 21487248
STAG2 STAG1 leukemia, sarcoma, glioblast-
oma and bladder cancer
Synthetic lethality 28430577
BRAF KRAS Colorectal adenocarcinoma;
lung cancer; ovary cancer;
gastric cancer; melanoma;
thyroid cancer; pancreas
cancer; cholangiocarcinoma
MAPK–ERK pathway 21102258; 20645028; 16721043;
19694828; 24959217;
23625203; 26996308;
27823638
PTEN PIK3CA Sarcoma; head and neck can-
cer; breast cancer; brain can-
cer; prostate cancer;
colorectal adenocarcinoma
PI3-kinase/AKT signal-
ing pathway
12569555; 22020193; 18097548;
19443396; 15805248;
ERBB2 KRAS Gastric cancer; lung cancer;
ovary cancer
EGFR/KRAS/BRAF
pathway
22315472; 25047676; 15753357;
17459062; 22899400
CDKN2A TP53 Leukemia; head and neck can-
cer; cholangiocarcinoma
Cell cycle pathway 15896902; 10675493; 18246599;
27495988
ERBB2 EGFR Lung cancer; gastric cancer;
head and neck cancer; brain
cancer
EGFR-RAS-RAF signaling
pathway
25047676; 22042947; 23855785;
17459062; 18757405
BRAF NRAS Thyroid cancer; brain cancer;
melanoma; leukemia; colo-
rectal adenocarcinoma
EGFR-RAS-RAF signaling
pathway
26575603; 21828154; 25576899;
24335104; 18381570;
25848750; 23860532;
16784981
Mutual exclusivity across cancer genomes | 3
剩余12页未读,继续阅读
2021-04-07 上传
2021-04-01 上传
点击了解资源详情
点击了解资源详情
点击了解资源详情
点击了解资源详情
点击了解资源详情
点击了解资源详情
点击了解资源详情

weixin_38640794
- 粉丝: 4
- 资源: 942
 我的内容管理
展开
我的内容管理
展开







最新资源
- 探索数据转换实验平台在设备装置中的应用
- 使用git-log-to-tikz.py将Git日志转换为TIKZ图形
- 小栗子源码2.9.3版本发布
- 使用Tinder-Hack-Client实现Tinder API交互
- Android Studio新模板:个性化Material Design导航抽屉
- React API分页模块:数据获取与页面管理
- C语言实现顺序表的动态分配方法
- 光催化分解水产氢固溶体催化剂制备技术揭秘
- VS2013环境下tinyxml库的32位与64位编译指南
- 网易云歌词情感分析系统实现与架构
- React应用展示GitHub用户详细信息及项目分析
- LayUI2.1.6帮助文档API功能详解
- 全栈开发实现的chatgpt应用可打包小程序/H5/App
- C++实现顺序表的动态内存分配技术
- Java制作水果格斗游戏:策略与随机性的结合
- 基于若依框架的后台管理系统开发实例解析
