GROMACS-2018分子模拟教程系列:入门与进阶
需积分: 5 22 浏览量
更新于2024-06-14
收藏 12.05MB PDF 举报
本文档是一份针对GROMACS-2018分子模拟软件的系列教程——"ALiveCoMSTutorial"。该教程由Justin A. Lemkul撰写,发表于2019年1月2日,旨在帮助用户深入了解并掌握分子动力学(MD)模拟技术在研究各种分子系统中的应用。MD模拟作为一种流行的原子级行为研究工具,对于想要从事这一领域的人来说,不仅需要对所选软件如GROMACS有深入理解,包括命令行操作、选项配置以及文件格式,还必须具备一定的物理学、数学知识,特别是计算机编程技能,并能够适应命令行环境。
教程内容涵盖了七个核心主题,这些主题旨在降低入门门槛,解决新用户在开始MD模拟时可能遇到的复杂性。每个教程都围绕实际操作和理论知识相结合,通过实例演示如何设置模拟参数、处理系统初始构型、执行动力学模拟、分析结果等关键步骤。例如,"FromProteinstoPerturbed Hamiltonians"部分可能着重于如何模拟蛋白质的动态行为,以及如何引入扰动来研究系统响应。
此外,文档强调了在线资源的重要性,提供了GitHub上的维护版本(<https://github.com/jalemkul/gmx_tutorials_livecoms>),用户可以通过这个平台提交反馈、建议或参与到教程的改进过程中。通过这种方式,作者鼓励互动与学习共享,确保教程内容的实用性和时效性。
GROMACS-2018的ALiveCoMSTutorial是一份全面且实用的指南,旨在帮助读者熟悉MD模拟的基础,并通过一系列系统的教学,提升他们在GROMACS软件上的操作能力和对分子动力学原理的理解。对于想要进入MD领域或提高现有技能的科研人员来说,这份教程是不可或缺的学习资料。

A LiveCoMS Tutorial
emstep = 0.01
nsteps = 50000
These instructions tell the GROMACS program
mdrun
(dis-
cussed below) to use the steepest descents method for en-
ergy minimization, and to terminate the process if the magni-
tude of the potential energy gradient is 1000.0 kJ mol
-1
nm
-1
or smaller. The maximum step size along the gradient is 0.01
nm, and a maximum of 50000 steps are allowed. The default
value of
emstep
in GROMACS of 0.01 nm is used, but it is often
necessary to use a smaller maximum step (e.g. 0.002 nm)
for systems that have difficulty converging; the smaller step
size allows for a more thorough walk over the potential en-
ergy surface, whereas a larger step may miss a path to the
true minimum. It is also possible to allow the process to go
on indefinitely, stopping only when convergence is reached
(due to numerical precision or by actually achieving
emtol
) by
setting
nsteps = -1
. Note that
emstep
has units of distance,
not time. Energy minimization is not a dynamical process;
time does not elapse between each step and there are no
velocities. Energy minimization steps are expressed in terms
of maximum displacement along the vector indicated by the
force.
The remainder of the
.mdp
file contains instructions for
how to compute nonbonded interactions:
ns tlist = 1
cutoff - sche me = Verlet
ns _type = grid
cou l o m b type = PME
rc oulomb = 1.0
rvdw = 1.0
pbc = xyz
The value of
nstlist
controls how many steps (minimization
or MD integration) elapse between updating the neighbor list
for determining which atoms contribute to the short-range
forces. In MD simulations, this value is larger because atoms
exchange from the neighbor list in diffusion-limited time, but
for energy minimization, it needs to be set to 1 because
the configuration may change considerably between each
step. The
cutoff-scheme = Verlet
setting uses a buffered
neighbor list, that is, atoms outside the longest cutoff are still
tracked to improve energy conservation. Neighbor searching
is done by checking atoms in neighboring grids (
ns_type =
grid
) rather than checking every possible atom (
ns_type =
simple
). All short-range nonbonded interactions (electrostat-
ics and van der Waals) are truncated at 1.0 nm (
rcoulomb =
1.0
and
rvdw= 1.0
), and periodic boundary conditions are
applied in all three dimensions (
pbc = xyz
). The PME method
is used to calculate long-range electrostatic forces.
Note that in
.mdp
files, there is no difference between
a hyphen (-) and underscore (_); hence in the above exam-
ple,
ns-type
and
ns_type
would be equivalent, as would
cutoff-scheme and cutoff_scheme.
As above, invoke
grompp
to assemble the instructions for
energy minimization (-f minim.mdp), coordinates
(
-c 1AKI_solv_ions.gro
), and topology (
-p topol.top
) to
create the run input file for energy minimization (
-o em.tpr
):
$ gmx gromp p -f minim . mdp -c
1 A K I _ s o l v _ i o n s . gro -p topol . top -o
em . tpr
The GROMACS
mdrun
program is responsible for perform-
ing all minimization and dynamics processes. To run energy
minimization, use the following syntax:
$ gmx mdrun -v - deffnm em
The
-v
option invokes "verbose" mode, in which an estimate
of time remaining is printed to the screen, along with the
current energy minimization step, the step size, potential
energy, and maximum force. It is not a required option, and if
printed to a file, may result in a large file that is saved to disk.
It is, however, instructive to watch the progress of energy
minimization to understand what is going on. The
-deffnm
option defines the base file name for all input and output
files and eliminates the need for using individual input and
output flags to specify names.
mdrun
requires the
.tpr
file as its only input, normally
passed to the
-s
flag.
mdrun
produces several file types, in-
cluding an ASCII text file with a
.log
extension, a binary file
with all energy values with a
.edr
extension, and trajectory
files with either
.trr
(full-precision coordinates, velocities,
and/or forces) or
.xtc
(reduced precision coordinates only)
extensions. When using
-deffnm
, these files will be called
em
,
with a corresponding file extension. It is useful to name files
in this manner to avoid relying on default file names (
md.log
,
ener.edr
,
traj.trr
,
traj_comp.xtc
), which will be the same
for any process carried out by mdrun.
When
mdrun
is done, the user will see something similar
to the following:
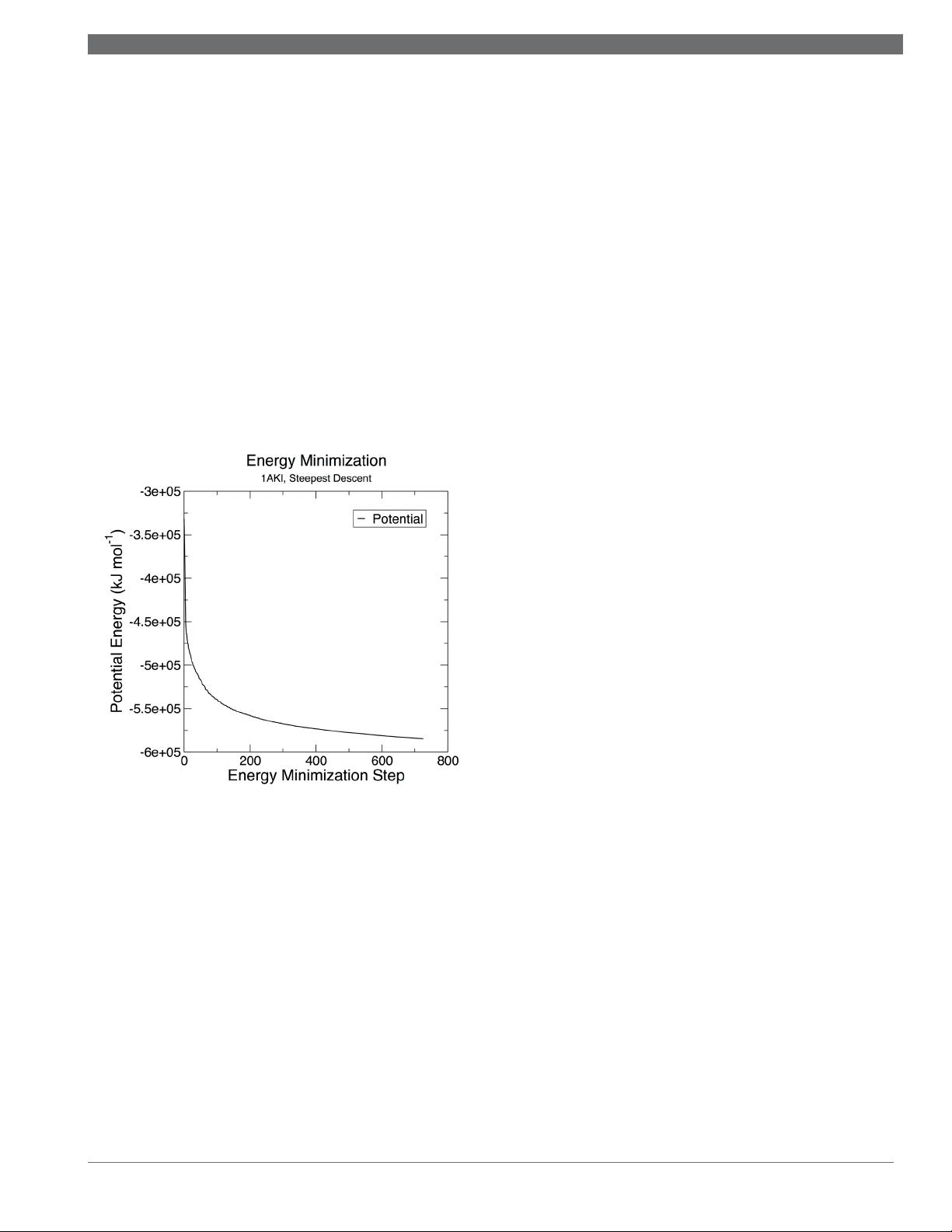
Stee p est D e scen t s co n verg e d to Fmax < 1000 i n 726 step s
Pote n tia l Ene r g y = -5. 8 4 4 8 7 69 e +05
Maxim u m force = 9.69 5 7593 e +02 on atom 736
Norm of f o r c e = 2 . 3290 9 28 e +01
For a system such as this, it is expected that the final po-
tential energy will be a negative value, on the order of 10
5
- 10
6
kJ mol
-1
. Potential energy is an extrinsic quantity, so
larger systems will have larger magnitudes. For a solvated
protein, the value will be negative, as favorable electrostatic
interactions between water molecules dominate the poten-
tial energy terms. Smaller systems may even have positive
potential energy, arising from the fact that in such cases, the
intramolecular bonded terms have a larger magnitude than
9 of 53
https://doi.org/10.33011/livecoms.1.1.5068
Living J. Comp. Mol. Sci. 2019, 1(1), 5068

剩余52页未读,继续阅读
点击了解资源详情
147 浏览量
261 浏览量
2024-05-21 上传
2019-09-26 上传
2023-06-20 上传
103 浏览量
136 浏览量

FrontScience
- 粉丝: 0
- 资源: 74
 我的内容管理
展开
我的内容管理
展开







最新资源
- 常见Windows 系统命令集合.txt
- JSP数据库编程指南
- JAVA配置文件编写说明文档
- Structs 文档
- Apress.Pro.LINQ.Language.Integrated.Query.in.C.Sharp.2008.Nov.2007
- CodeSmith开发资料
- Apress.Pro.C.Sharp.2008.and.the.dot.NET.3.5.Platform.4th.Edition.Nov.2007
- C#读写INI文件(Word)
- java 编程 思想.[[書籍][圖書]电子书].pdf
- Apress.Pro.C.Sharp.2005.and.the.dot.NET.2.0.Platform.3rd.Edition.Sep.2005
- 程序员考试模拟试卷.doc
- 2008年程序员考试模拟试卷
- Apress.Expert.Service.Oriented.Architecture.in.C.Sharp.2005.2nd.Edition.Aug.2006
- linux的c入门.pdf
- Absolute C++英文版
- Apress.Accelerated.C.Sharp.2008.Nov.2007
