LAMMPS分子模拟软件详解及可视化工具推荐
需积分: 40 144 浏览量
更新于2024-07-15
1
收藏 2.8MB PDF 举报
LAMMPS个人注释
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一款在计算材料科学领域广泛应用的通用分子模拟软件,因其高度的灵活性、高效性和可扩展性而受到广泛赞誉。这款开源软件支持多种势能模型,使得它能够在模拟软材料和固体物理系统时展现出卓越性能。LAMMPS的特点包括并行化处理能力,易于理解和扩展的编程接口,以及丰富的文档支持。
在软件下载部分,有多个实用工具可供选择。例如,对于原子结构的可视化,用户可以选择Linux下的大规模原子可视化软件atomeye,它专为大规模原子系统设计,提供直观的图形界面。VMD(Visual Molecular Dynamics)则是一个功能全面的分子可视化程序,适用于生物大分子系统的展示、动画和分析,其内置脚本功能使其具备高度定制化能力。另外,XCrySDen是一个晶体和分子结构可视化工具,适用于显示等值面和轮廓,能在多种UNIX平台上运行,无需特殊硬件配置。
在计算程序方面,LAMMPS是核心推荐之一,作为强大的分子动力学(MD)代码,它具有高性能、易用性和广泛的社区支持。Gromacs和DL-POLY也是MD模拟的优秀选项,分别以其易用性和多功能性著称,适合进行经典分子动力学模拟。NAMD则针对大型生物分子系统的模拟设计,追求高性能计算。
对于那些依赖于第一性原理(如密度泛函理论,DFT)的ab initio计算,VASP是一个备受推崇的选择,它的功能强大且用户界面友好,被广泛用于材料科学研究中的电子结构计算。
LAMMPS个人注释提供了丰富的资源,涵盖了从可视化工具到高级计算程序的完整链条,满足不同层次的用户需求,无论是初学者还是专业研究人员都能从中受益。通过这些软件,研究者能够深入探索材料的微观行为,预测和优化新材料性能,推动科技前沿的发展。
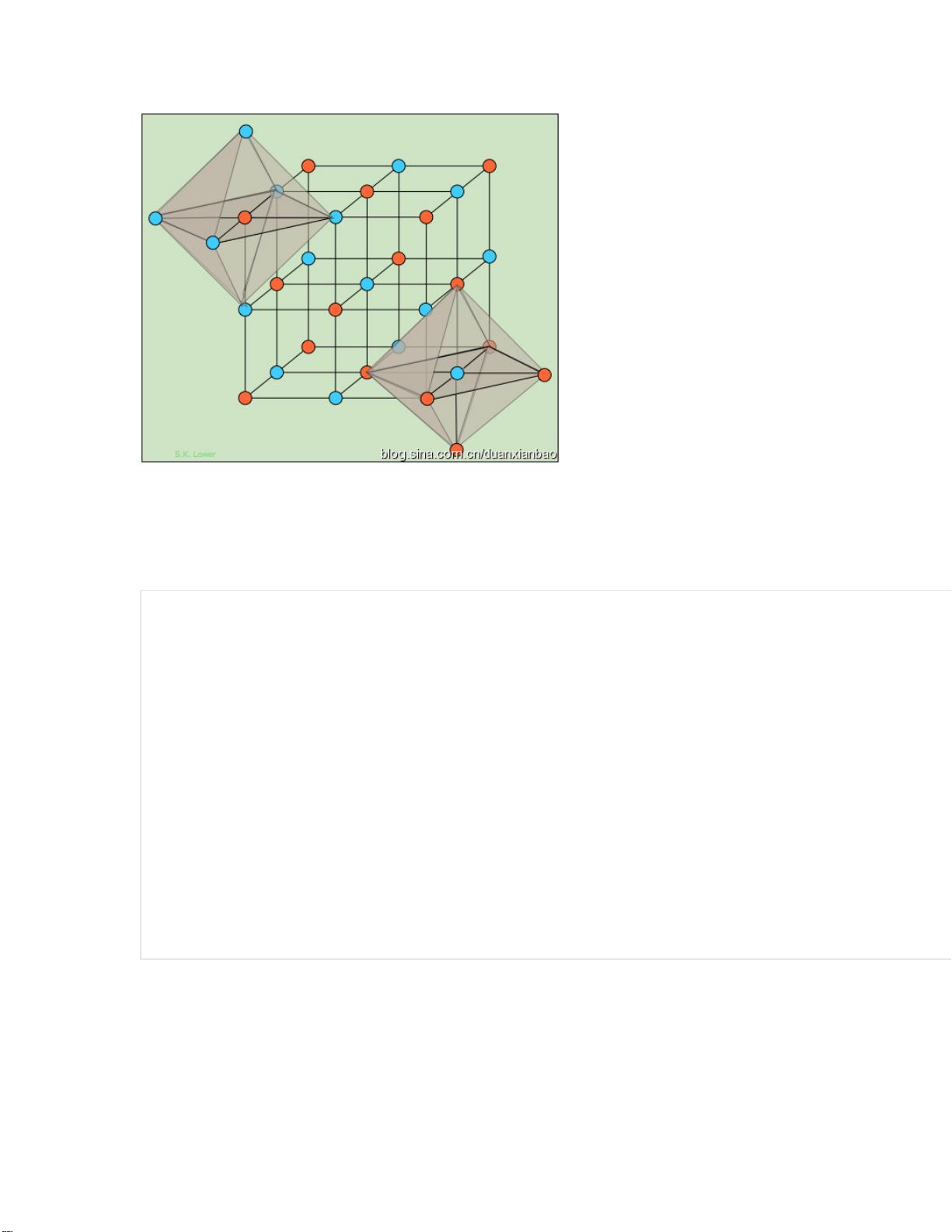
创建结构的命令不外乎 lattice, create_box, create_atoms,这里只是强调在合金体
系创建的应用。
先直接给出命令,如下:
01
02
03
04
05
06
07
lattice custom $x a1 1.0 0.0 0.0 a2 0.0 1.0 0.0 a3 0.0 0.0 1.0 &
basis
0.0 0.0 0.0 basis 0.5 0.5 0.0 basis 0.5 0.0 0.5 b
basis
0.5 0.5 0.5 basis 0.0 0.0 0.5 basis 0.0 0.5 0.0 b
region box block 0 3 0 3 0 3
create_box 2 box
create_atoms 2 box basis 1 1 basis 2 1 basis 3 1 basis 4 1 &
basis 5 2 basis 6 2 basis 7 2 basis 8 2
具体说明:
(1) lattice 第一行为晶格矢量,其中$x 为晶格矢量;第二行和第三行每一个 basis
对应原胞中的一个原子。对于 B1 结构,是包含 8 个原子,即 4 个 Na,4 个 Cl。
(2) region 是定义盒子的区域


剩余160页未读,继续阅读
2023-06-28 上传
2023-12-26 上传
2023-03-16 上传
2023-07-16 上传
2024-01-03 上传
2023-11-03 上传

qq95483431
- 粉丝: 8
- 资源: 19
 我的内容管理
展开
我的内容管理
展开







最新资源
- Hadoop生态系统与MapReduce详解
- MDS系列三相整流桥模块技术规格与特性
- MFC编程:指针与句柄获取全面解析
- LM06:多模4G高速数据模块,支持GSM至TD-LTE
- 使用Gradle与Nexus构建私有仓库
- JAVA编程规范指南:命名规则与文件样式
- EMC VNX5500 存储系统日常维护指南
- 大数据驱动的互联网用户体验深度管理策略
- 改进型Booth算法:32位浮点阵列乘法器的高速设计与算法比较
- H3CNE网络认证重点知识整理
- Linux环境下MongoDB的详细安装教程
- 压缩文法的等价变换与多余规则删除
- BRMS入门指南:JBOSS安装与基础操作详解
- Win7环境下Android开发环境配置全攻略
- SHT10 C语言程序与LCD1602显示实例及精度校准
- 反垃圾邮件技术:现状与前景
