




LAMMPS学习与应用指南
需积分: 45 98 浏览量
更新于2024-07-17
10
收藏 2.8MB PDF 举报
"lammps使用手册,这是一份自我学习的lammps学习资料,包含了网络上各种信息的大杂烩,旨在保证内容的完整性未做修改。这份手册特别适用于想要深入理解和应用lammps的人群。"
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一种强大的分子动力学(MD)模拟软件,它被设计成高效、并行化且文档详尽,同时易于扩展和开源。LAMMPS广泛应用于材料科学、化学、物理以及工程领域的研究,能够模拟从纳米到微米尺度的系统,包括固体、液体、气体和生物分子等。
LAMMPS的特点在于其模块化设计,允许用户添加新的计算方法和力场。此外,LAMMPS支持多种并行计算策略,如MPI(消息传递接口)和OpenMP,使得在多核处理器或分布式计算集群上运行大规模模拟成为可能。通过内置的脚本语言,用户可以编写复杂的模拟流程,实现自动化模拟和数据后处理。
在学习LAMMPS时,推荐使用以下辅助资源:
1. 可视化软件:
- AtomEye:专为Linux设计的大型原子结构可视化工具。
- VESTA:用户友好的可视化软件,适合初学者。
- VMD:分子可视化程序,用于展示、动画和分析大型生物分子系统。
- XCrySDen:晶体和分子结构可视化程序,支持等值面和轮廓显示,并能叠加在晶体结构上进行交互操作。
2. 计算程序:
- GROMACS:另一个优秀的免费MD软件,特别适合生物分子系统的模拟。
- DL-POLY:一款通用的经典分子动力学模拟软件,开源。
- NAMD:针对大型生物分子系统设计的并行MD代码,性能强大。
3. 首次接触LAMMPS,你需要理解基本概念,如分子动力学的基本原理、力场的选择和设置、时间步长的选取、边界条件等。然后,你可以通过阅读LAMMPS的手册和示例来熟悉命令行界面和输入文件的结构。此外,LAMMPS社区提供了丰富的教程和案例,帮助用户快速上手。
4. 在进行实际模拟之前,确保你了解所选势函数的适用范围,因为不同的势函数会直接影响模拟结果的准确性。例如,EAM(嵌入式原子模型)、LJ(Lennard-Jones)势、CHARMM、AMBER等都是常见的势函数类型。
5. 数据分析是LAMMPS模拟过程中的重要环节,通常涉及计算系统能量、压力、扩散系数、结构因子等。LAMMPS内建了一些数据分析工具,也可以结合其他科学分析软件如GROMACS的gmx tools或VMD的分析插件进行深入分析。
LAMMPS作为材料科学中的强大工具,需要结合合适的可视化和计算软件进行综合使用。通过深入学习和实践,你可以充分利用其功能,解决各种复杂的模拟问题。记得在学习过程中,利用在线资源和社区支持,这将对你的研究大有裨益。
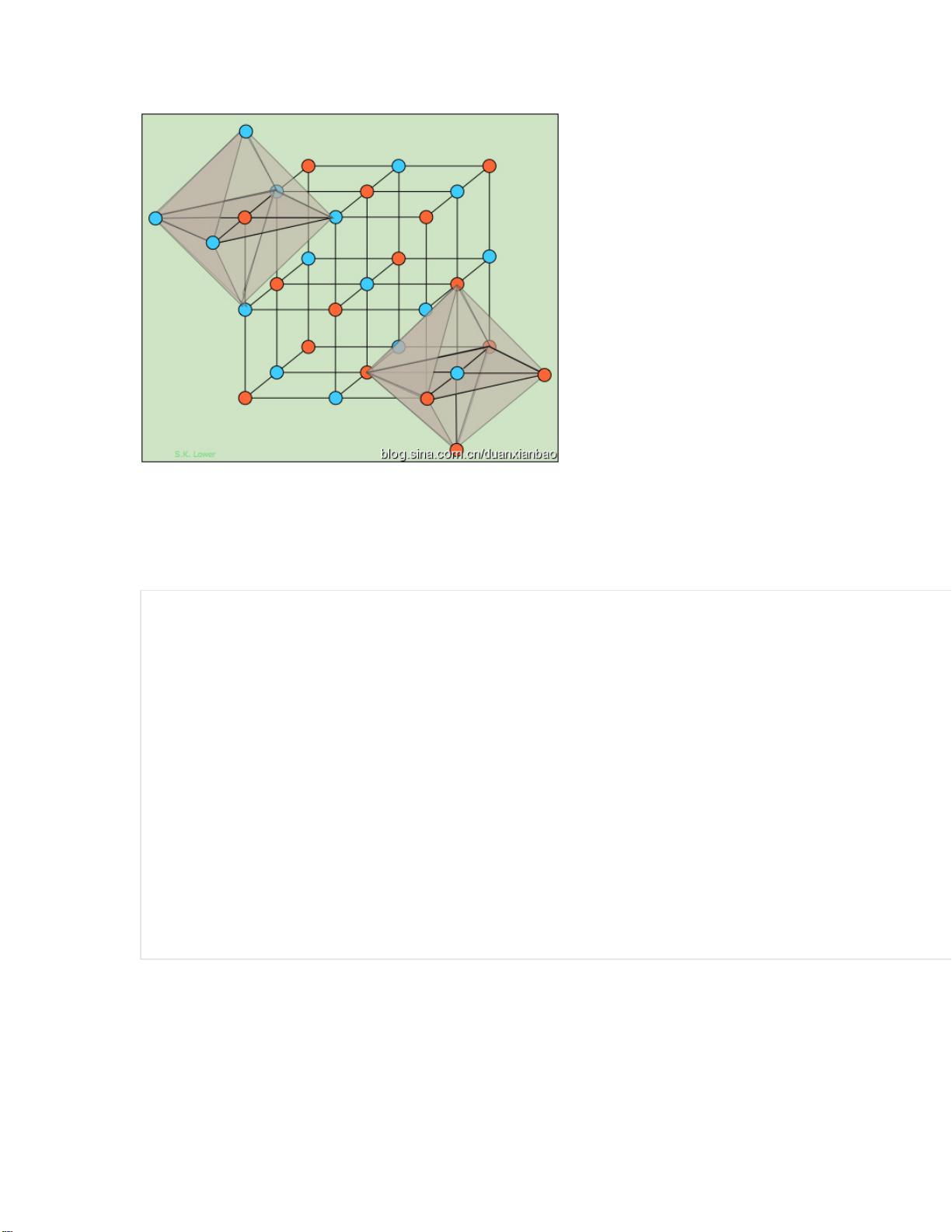
创建结构的命令不外乎 lattice, create_box, create_atoms,这里只是强调在合金体
系创建的应用。
先直接给出命令,如下:
01
02
03
04
05
06
07
lattice custom $x a1 1.0 0.0 0.0 a2 0.0 1.0 0.0 a3 0.0 0.0 1.0 &
basis
0.0 0.0 0.0 basis 0.5 0.5 0.0 basis 0.5 0.0 0.5 b
basis
0.5 0.5 0.5 basis 0.0 0.0 0.5 basis 0.0 0.5 0.0 b
region box block 0 3 0 3 0 3
create_box 2 box
create_atoms 2 box basis 1 1 basis 2 1 basis 3 1 basis 4 1 &
basis 5 2 basis 6 2 basis 7 2 basis 8 2
具体说明:
(1) lattice 第一行为晶格矢量,其中$x 为晶格矢量;第二行和第三行每一个 basis
对应原胞中的一个原子。对于 B1 结构,是包含 8 个原子,即 4 个 Na,4 个 Cl。
(2) region 是定义盒子的区域


剩余160页未读,继续阅读
569 浏览量
2237 浏览量
194 浏览量
3286 浏览量
378 浏览量
159 浏览量
209 浏览量
1044 浏览量
2462 浏览量

weixin_42252074
- 粉丝: 1
 我的内容管理
展开
我的内容管理
展开







最新资源
- PHP中快速生成二维码的技巧和工具
- Python程序pspstc有效解决固定导热系数问题
- C#初学者必备:dll使用案例解析
- 全面解析NorFlash与NandFlash差异对比
- Micron.js:炫酷CSS3动画的JavaScript动画库插件
- 360文档下载神器:轻松获取积分限制内容
- 初学者指南:掌握Android小程序的经典布局与ListView
- PageAdmin建站系统:强大的企业级.NET网站管理平台
- Excel合同审批表模板(新)高效办公必备
- 金鑫影像工作站:图像采集与处理专家
- 便捷安全的绿色版即时截屏工具
- 自学eclipse开发:JFrame实现Hello World程序
- JavaScript代码拆分实践与优化指南
- 计算机英语第三版课程习题及答案解析
- 欧美风电脑主机HTML模板下载
- ARM开发板BootLoader深入分析与技术内幕






















