入门指南:Materials Studio与Castep的DFT计算与硅晶格模拟
需积分: 4 168 浏览量
更新于2024-07-31
收藏 3.12MB PDF 举报
本指南是一份针对初学者的全面教程,介绍了如何在Materials Studio中使用CASTEP进行密度泛函理论(DFT)计算。CASTEP是一种广泛应用的量子力学软件,用于模拟材料的电子结构和动力学性质。本文档将逐步引导你创建一个新的Materials Studio项目,并进行一个8个原子硅的体心立方(FCC)晶格模拟。
首先,你需要在Materials Studio中创建一个新的项目,这将作为所有计算文件的容器。接着,你将在“3D Atomistic文档”中设置模拟单元。对于硅晶体,其空间群是FD3M,代表钻石结构,具有非对称性。通过告知Materials Studio硅的对称性,它会自动应用这些信息到原子上,使原子位于相应的对称点上。
在构建晶格部分,点击“Lattice”选项卡,因为Materials Studio只支持立方体(FCC)晶格,所以只需要输入一个参数——立方晶胞的边长,这里设置为5.4 Å。点击“Build”按钮后,晶格结构即被定义。
下一步是添加硅原子。在“Build Crystal”窗口中,将元素更改为硅,然后点击“Add”键,将硅原子置于晶格原点。这样就成功地设置了模拟的基础结构。
DFT计算的核心在于设置计算参数,如交换关联势、自洽场迭代等,这部分通常在项目设置或计算模块中完成。对于初学者来说,可能需要了解基本的DFT概念,例如Kohn-Sham方程,以及如何选择合适的交换-correlation函数(如LDA或GGA)。此外,可能还需要设置计算网格密度,以确保结果的精度。
在模拟过程中,你将导出并分析计算结果,如电子密度、能带结构、态密度等,这些都是理解材料性质的关键。同时,可能需要学习如何解读和可视化这些数据,以便于理解和解释你的模拟。
最后,要注意,随着经验的增长,你可能需要处理更大规模的系统、更复杂的材料,或者考虑温度、压力等外部因素对材料的影响。这时,熟悉Materials Studio的高级功能,如并行计算和自动化脚本编写,将大大提高效率。
总结来说,这篇指南为你提供了一个从零开始学习Materials Studio和CASTEP结合进行DFT计算的步骤,包括单元晶格构造、对称性设置、参数调整以及初步的数据分析。通过实践和深入理解,你将能够熟练地运用这些工具来探索和预测材料的性质。
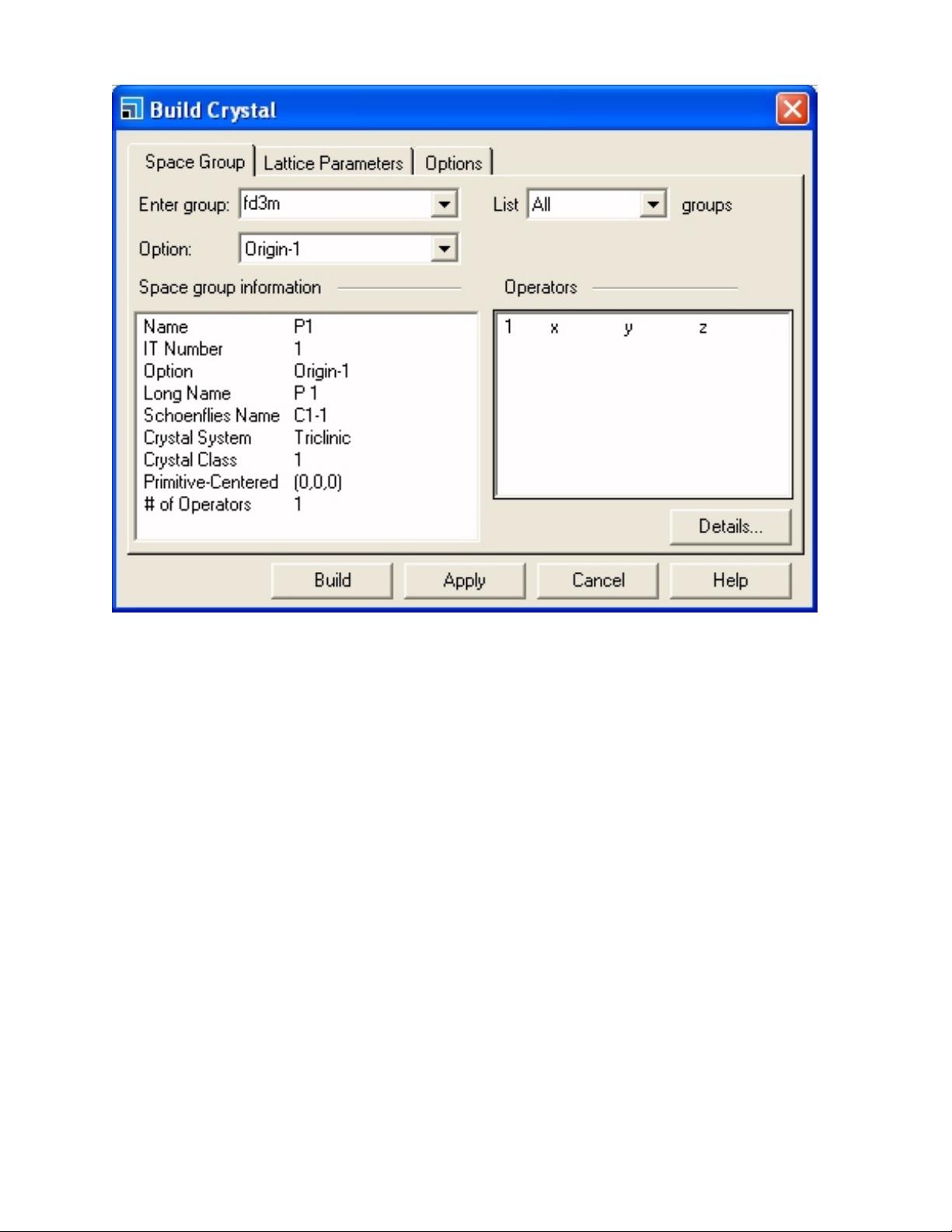
The default is space group P1, i.e. no symme-
try. Silicon has the diamond structure (space
group FD3M). By telling Materials Studio this
symmetry it will automatically apply it to the
atoms, thus generating atoms at the symmetry
points.
6

剩余37页未读,继续阅读
2023-03-29 上传
2023-03-31 上传
2023-06-03 上传
2023-03-31 上传
2023-03-16 上传
2023-06-13 上传
2023-04-21 上传
2023-04-10 上传
2023-05-22 上传

fn001cn
- 粉丝: 16
- 资源: 59
 我的内容管理
展开
我的内容管理
展开







最新资源
- 构建Cadence PSpice仿真模型库教程
- VMware 10.0安装指南:步骤详解与网络、文件共享解决方案
- 中国互联网20周年必读:影响行业的100本经典书籍
- SQL Server 2000 Analysis Services的经典MDX查询示例
- VC6.0 MFC操作Excel教程:亲测Win7下的应用与保存技巧
- 使用Python NetworkX处理网络图
- 科技驱动:计算机控制技术的革新与应用
- MF-1型机器人硬件与robobasic编程详解
- ADC性能指标解析:超越位数、SNR和谐波
- 通用示波器改造为逻辑分析仪:0-1字符显示与电路设计
- C++实现TCP控制台客户端
- SOA架构下ESB在卷烟厂的信息整合与决策支持
- 三维人脸识别:技术进展与应用解析
- 单张人脸图像的眼镜边框自动去除方法
- C语言绘制图形:余弦曲线与正弦函数示例
- Matlab 文件操作入门:fopen、fclose、fprintf、fscanf 等函数使用详解
