Amber软件动力学模拟指南:从能量最小化到实际模拟
版权申诉
96 浏览量
更新于2024-08-06
1
收藏 1.29MB PDF 举报
"Amber软件是分子动力学模拟的一个强大工具,主要用于研究生物大分子如蛋白质和核酸的动力学行为。该软件通过解决牛顿运动方程来模拟分子系统的动态过程。在Amber中,动力学模拟通常包括三个主要步骤:能量最小化、体系平衡和生产运行。
1) 能量最小化:这是模拟的第一步,目的是消除模型结构中的内应力。通过imin=1参数设定,Amber将执行能量最小化任务。ntx=1表示读取坐标信息,而nstep参数设定了最小化迭代次数,用于控制最小化过程的持续时间。
2) 体系平衡:在能量最小化后,系统需要被加热和膨胀至模拟的生理条件,以达到热力学平衡。这通常涉及恒温-恒压(NPT)或恒温(NVT) Ensemble的模拟。关键参数如nteq和ncyc用于设置平衡阶段的迭代次数,以确保系统稳定。
3) 生产运行:当体系达到平衡后,便开始进行实际的动力学模拟,收集数据以分析分子动力学行为。这部分通常需要较长的时间,并且可以通过istep参数设定采样间隔。
在Amber模拟过程中,用户需要准备参数文件(prmtop)、坐标文件(inpcrd)以及sander程序的配置文件(mdin)。配置文件mdin中包含了一系列键/值对,用于设定模拟的具体条件,如温度(temp)、压力(press)、时间步长(dt)等。此外,sander程序运行时还会生成临时的rst文件记录坐标和速度,以及轨迹文件(mdcrd)保存模拟过程的原子坐标。
在进行动力学模拟时,用户还需要考虑其他的细节,比如使用合适的力场(force field)来描述分子间的相互作用,选择合适的边界条件(如PBC,周期性边界条件),以及应用适当的约束,如氢键固定(例如使用iwrap和_constraints选项)。
Amber软件提供了全面的功能来实现复杂生物分子的动力学模拟,但正确配置和理解每一个模拟步骤及参数至关重要,这对于获取可靠的模拟结果和深入的生物学见解是必不可少的。"
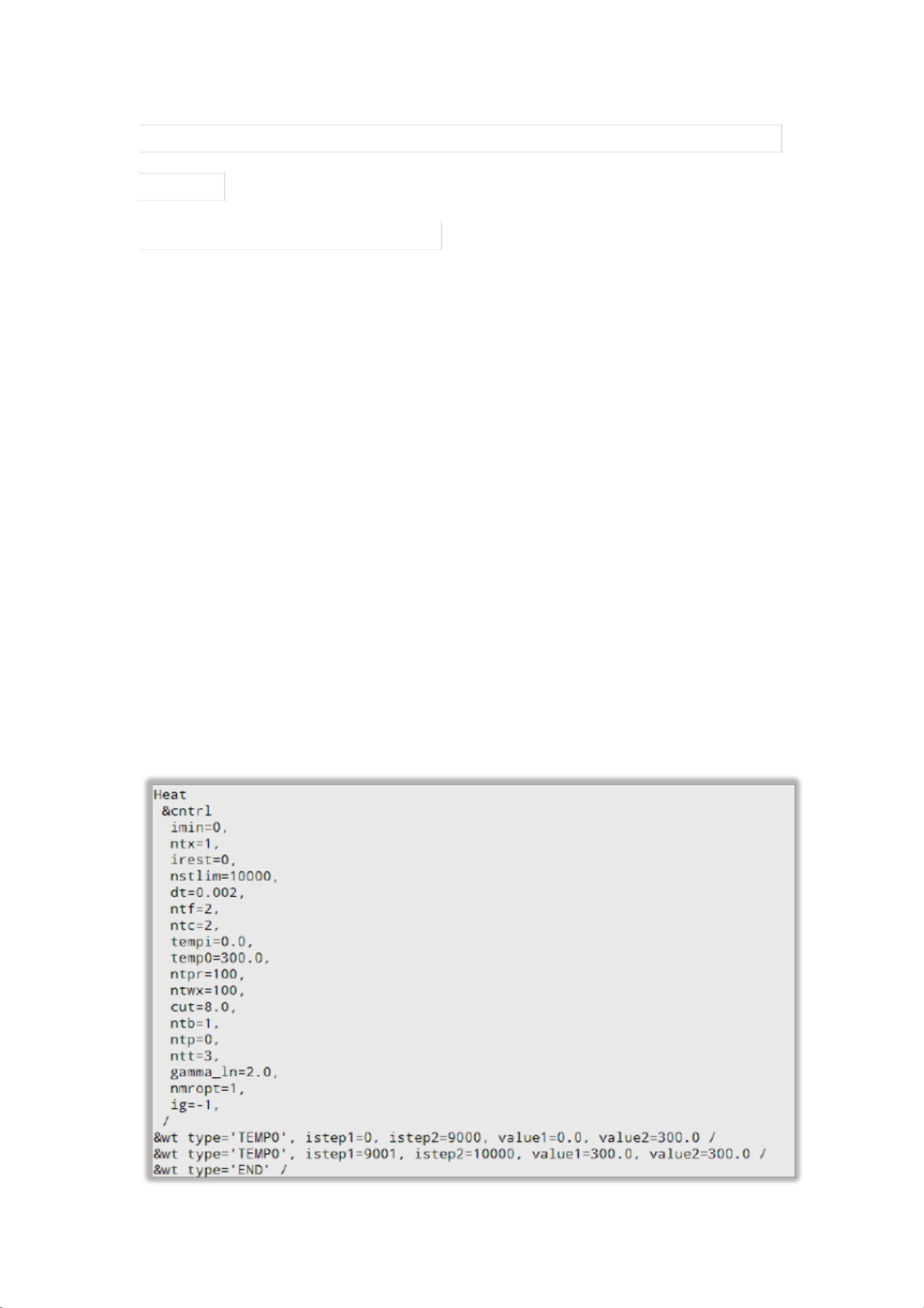
第一行是标题, &cntrl 是起始符, ”/ ”是结束符, 中间的键 /值对就是参
数配置。
上述的参数配置可以归纳如下:
imin=1 Choose a minimization run,指定做能量最小化的动力学。
ntx=1
Read coordinates but not velocities from ASCII formatted inpcrd coordinate file
ntx 指定如何获得坐标信息,这里直接从 inpcrd 中读取坐标信息。
maxcyc=2000 Maximum minimization cycles
能量最小化的算法涉及循环迭代,这里指定迭代次数。
ncyc=1000 The steepest descent algorithm for the first 0-ncyc cycles, then switches the
conjugate gradient algorithm for ncyc-maxcyc cycles
如上所说,循环迭代的算法不同,此处指定到哪一步第一种迭代算法结
束。
ntpr=100 Print to the Amber mdout output file every ntpr cycles
这个表明采集计算信息的频率,输出到 mdout 文件中。
cut=8.0
Nonbonded cutoff distance in Angstroms
由于计算能量时需要有一个截断距离,这个参数指定截断距离。
其余两个参数都是能量最小化时不可用的参数,下面会具体说明。
2)Heating ( Equilibrium step1 )
剩余13页未读,继续阅读
2022-03-05 上传
2021-10-19 上传
2021-11-08 上传
2024-03-28 上传
2020-07-14 上传
2022-09-23 上传
2022-10-20 上传
2019-10-04 上传
点击了解资源详情
2024-10-24 上传

hyh15959933972
- 粉丝: 0
- 资源: 8万+
 我的内容管理
展开
我的内容管理
展开







最新资源
- 掌握Jive for Android SDK:示例应用的使用指南
- Python中的贝叶斯建模与概率编程指南
- 自动化NBA球员统计分析与电子邮件报告工具
- 下载安卓购物经理带源代码完整项目
- 图片压缩包中的内容解密
- C++基础教程视频-数据类型与运算符详解
- 探索Java中的曼德布罗图形绘制
- VTK9.3.0 64位SDK包发布,图像处理开发利器
- 自导向运载平台的行业设计方案解读
- 自定义 Datadog 代理检查:Python 实现与应用
- 基于Python实现的商品推荐系统源码与项目说明
- PMing繁体版字体下载,设计师必备素材
- 软件工程餐厅项目存储库:Java语言实践
- 康佳LED55R6000U电视机固件升级指南
- Sublime Text状态栏插件:ShowOpenFiles功能详解
- 一站式部署thinksns社交系统,小白轻松上手
