SampleID=JN_1901 Blast_out=/Analysis_B/20220326_JNILI/006_blastx/ Diamond_Refer="/home/Reference_database/Diamond_blastx_nr/Diamond_nr.dmnd" Diamond_file_in=/Analysis_B/20220326_JNILI/004_Trinity/JN_1901_trinity.fa #### 003 #### cd ${Blast_out} source activate yerunze_diamond diamond blastx -d ${Diamond_Refer} -p 16 -q ${Diamond_file_in} -o ${SampleID}_blastx --outfmt 6 qtitle qlen sseqid pident length qstart qend evalue bitscore staxids stitle salltitles sscinames -e 1e-5 -k 1 -b5 -c1这段代码什么意思
时间: 2024-02-19 07:00:40 浏览: 132
这是一段基于Diamond软件进行blastx比对的代码。Diamond是一款高效的序列比对软件,可以在较短时间内完成大规模序列比对任务。
具体来说,这段代码的作用是对输入的JN_1901_trinity.fa文件进行blastx比对,比对的参考数据库为Diamond_nr.dmnd。比对结果将以outfmt 6的格式输出到文件JN_1901_blastx中,包括查询序列ID、长度、匹配的参考序列ID、匹配度、匹配长度、查询序列起始和终止位置、E值、比特分数、参考序列分类信息等。比对时使用了16个线程,设置了一些参数如-e 1e-5表示E值阈值为1e-5,-k 1表示输出最佳匹配,-b5表示输出最多5个相似序列,-c1表示输出序列分类信息。
相关问题
SampleID=JN_1901 Blast_out=/Analysis_B/20220326_JNILI/007_blastn/ Diamond_file_in=/Analysis_B/20220326_JNILI/004_Trinity/JN_1901_trinity.fa #### 003 #### cd ${Blast_out} blastn -query ${Diamond_file_in} -db "/home/Reference_database/NCBI_db_nt/" -out ${Blast_out}${SampleID}_blastn.out -num_threads 12 -outfmt 6 qseqid qlen saccver salltitles slen pident length qcovs bitscore evalue staxids -evalue 1e-5 awk -F$'/t' '!seen[$1]++' ${Blast_out}${SampleID}_blastn.out > ${Blast_out}${SampleID}_blastn.out.unique帮我解释一下这段代码
这段代码主要是进行基因序列比对和去重的过程,其中:
- SampleID=JN_1901 是样本的编号;
- Blast_out=/Analysis_B/20220326_JNILI/007_blastn/ 是blast比对结果的输出路径;
- Diamond_file_in=/Analysis_B/20220326_JNILI/004_Trinity/JN_1901_trinity.fa 是基因组装结果的路径;
- cd ${Blast_out} 进入blast比对结果的输出路径;
- blastn -query ${Diamond_file_in} -db "/home/Reference_database/NCBI_db_nt/" -out ${Blast_out}${SampleID}_blastn.out -num_threads 12 -outfmt 6 qseqid qlen saccver salltitles slen pident length qcovs bitscore evalue staxids -evalue 1e-5 进行blastn比对,-query指定比对的查询序列,-db指定参考序列库,-out指定输出文件名,-num_threads指定线程数,-outfmt指定输出格式,这里是tabular格式;
- awk -F$'/t' '!seen[$1]++' ${Blast_out}${SampleID}_blastn.out > ${Blast_out}${SampleID}_blastn.out.unique 根据比对结果中的qseqid(即查询序列的ID)进行去重,这里使用awk命令,-F指定分隔符为制表符,!seen[$1]++表示如果第一个字段(即qseqid)没有出现过,则保留该行,否则忽略该行,最终输出去重后的结果到${Blast_out}${SampleID}_blastn.out.unique文件中。
blastn -query ${Diamond_file_in} -db "/home/Reference_database/NCBI_db_nt/" -out ${Blast_out}${SampleID}_blastn.out -num_threads 12 -outfmt 6 qseqid qlen saccver salltitles slen pident length qcovs bitscore evalue staxids -evalue 1e-5 awk -F$'/t' '!seen[$1]++' ${Blast_out}${SampleID}_blastn.out > ${Blast_out}${SampleID}_blastn.out.unique帮我解析一下这段代码
这段代码是用于执行Blastn比对,并处理Blastn比对结果的操作。下面是它的详细解释:
1. `blastn -query ${Diamond_file_in} -db "/home/Reference_database/NCBI_db_nt/" -out ${Blast_out}${SampleID}_blastn.out -num_threads 12 -outfmt 6 qseqid qlen saccver salltitles slen pident length qcovs bitscore evalue staxids -evalue 1e-5`:这是Blastn的命令行,其中`${Diamond_file_in}`是输入的序列文件路径,`/home/Reference_database/NCBI_db_nt/`是参考数据库的路径,`${Blast_out}${SampleID}_blastn.out`是输出结果文件的路径和文件名,`-num_threads 12`表示使用12个线程进行比对,`-outfmt 6`指定输出格式为表格格式,接下来的各个参数指定了输出表格中每一列的信息。
2. `awk -F$'/t' '!seen[$1]++' ${Blast_out}${SampleID}_blastn.out > ${Blast_out}${SampleID}_blastn.out.unique`:这是用于处理Blastn比对结果的操作,其中`${Blast_out}${SampleID}_blastn.out`是Blastn比对输出的结果文件,`> ${Blast_out}${SampleID}_blastn.out.unique`表示将处理结果输出到`${Blast_out}${SampleID}_blastn.out.unique`文件中。`awk -F$'/t' '!seen[$1]++'`是一个awk命令,它的作用是将`${Blast_out}${SampleID}_blastn.out`文件中的重复行去除,只保留第一次出现的行。其中`-F$'/t'`指定了表格文件的分隔符为制表符,`!seen[$1]++`表示只输出第一次出现的行,去除重复行。
因此,这段代码的作用是将`${Diamond_file_in}`中的序列文件与参考数据库中的核酸序列进行比对,并将比对结果输出到`${Blast_out}${SampleID}_blastn.out`文件中,然后使用awk命令去除比对结果中的重复行,并将处理结果输出到`${Blast_out}${SampleID}_blastn.out.unique`文件中。
阅读全文
相关推荐
最新推荐
Blast序列比对与利用mega构建进化树
《Blast序列比对与利用mega构建进化树详解》 在生物信息学研究中,序列比对和进化树构建是两个关键步骤,它们对于理解基因或蛋白质的进化关系至关重要。本篇文章将详细介绍如何在国家生物技术信息中心(NCBI)上...
JavaScript实现的高效pomodoro时钟教程
资源摘要信息:"JavaScript中的pomodoroo时钟"
知识点1:什么是番茄工作法
番茄工作法是一种时间管理技术,它是由弗朗西斯科·西里洛于1980年代末发明的。该技术使用一个定时器来将工作分解为25分钟的块,这些时间块之间短暂休息。每个时间块被称为一个“番茄”,因此得名“番茄工作法”。该技术旨在帮助人们通过短暂的休息来提高集中力和生产力。
知识点2:JavaScript是什么
JavaScript是一种高级的、解释执行的编程语言,它是网页开发中最主要的技术之一。JavaScript主要用于网页中的前端脚本编写,可以实现用户与浏览器内容的交云互动,也可以用于服务器端编程(Node.js)。JavaScript是一种轻量级的编程语言,被设计为易于学习,但功能强大。
知识点3:使用JavaScript实现番茄钟的原理
在使用JavaScript实现番茄钟的过程中,我们需要用到JavaScript的计时器功能。JavaScript提供了两种计时器方法,分别是setTimeout和setInterval。setTimeout用于在指定的时间后执行一次代码块,而setInterval则用于每隔一定的时间重复执行代码块。在实现番茄钟时,我们可以使用setInterval来模拟每25分钟的“番茄时间”,使用setTimeout来控制每25分钟后的休息时间。
知识点4:如何在JavaScript中设置和重置时间
在JavaScript中,我们可以使用Date对象来获取和设置时间。Date对象允许我们获取当前的日期和时间,也可以让我们创建自己的日期和时间。我们可以通过new Date()创建一个新的日期对象,并使用Date对象提供的各种方法,如getHours(), getMinutes(), setHours(), setMinutes()等,来获取和设置时间。在实现番茄钟的过程中,我们可以通过获取当前时间,然后加上25分钟,来设置下一个番茄时间。同样,我们也可以通过获取当前时间,然后减去25分钟,来重置上一个番茄时间。
知识点5:实现pomodoro-clock的基本步骤
首先,我们需要创建一个定时器,用于模拟25分钟的工作时间。然后,我们需要在25分钟结束后提醒用户停止工作,并开始短暂的休息。接着,我们需要为用户的休息时间设置另一个定时器。在用户休息结束后,我们需要重置定时器,开始下一个工作周期。在这个过程中,我们需要为每个定时器设置相应的回调函数,以处理定时器触发时需要执行的操作。
知识点6:使用JavaScript实现pomodoro-clock的优势
使用JavaScript实现pomodoro-clock的优势在于JavaScript的轻量级和易学性。JavaScript作为前端开发的主要语言,几乎所有的现代浏览器都支持JavaScript。因此,我们可以很容易地在网页中实现pomodoro-clock,用户只需要打开网页即可使用。此外,JavaScript的灵活性也使得我们可以根据需要自定义pomodoro-clock的各种参数,如工作时间长度、休息时间长度等。
管理建模和仿真的文件
管理Boualem Benatallah引用此版本:布阿利姆·贝纳塔拉。管理建模和仿真。约瑟夫-傅立叶大学-格勒诺布尔第一大学,1996年。法语。NNT:电话:00345357HAL ID:电话:00345357https://theses.hal.science/tel-003453572008年12月9日提交HAL是一个多学科的开放存取档案馆,用于存放和传播科学研究论文,无论它们是否被公开。论文可以来自法国或国外的教学和研究机构,也可以来自公共或私人研究中心。L’archive ouverte pluridisciplinaire
【WebLogic客户端兼容性提升秘籍】:一站式解决方案与实战案例
# 摘要
WebLogic作为一款广泛使用的中间件产品,其客户端兼容性对于企业应用至关重要。本文从基本概念出发,系统地介绍了WebLogic的架构、组件以及兼容性问题的分类和影响。通过深入分析兼容性测试方法和诊断分析技术,探讨了如何有效地识别和解决客户端兼容性问题。进一步,本文提出了提升兼容性的策略,包括代码层面的设计、配置管理、补丁升级以及快速响应流程。最后,结合实战案例,本文详细说明了解决方案的实施过
使用jupyter读取文件“近5年考试人数.csv”,绘制近5年高考及考研人数发展趋势图,数据如下(单位:万人)。
在Jupyter Notebook中读取CSV文件并绘制图表,通常需要几个步骤:
1. 首先,你需要导入必要的库,如pandas用于数据处理,matplotlib或seaborn用于数据可视化。
```python
import pandas as pd
import matplotlib.pyplot as plt
```
2. 使用`pd.read_csv()`函数加载CSV文件:
```python
df = pd.read_csv('近5年考试人数.csv')
```
3. 确保数据已经按照年份排序,如果需要的话,可以添加这一行:
```python
df = df.sor
CMake 3.25.3版本发布:程序员必备构建工具
资源摘要信息:"Cmake-3.25.3.zip文件是一个包含了CMake软件版本3.25.3的压缩包。CMake是一个跨平台的自动化构建系统,用于管理软件的构建过程,尤其是对于C++语言开发的项目。CMake使用CMakeLists.txt文件来配置项目的构建过程,然后可以生成不同操作系统的标准构建文件,如Makefile(Unix系列系统)、Visual Studio项目文件等。CMake广泛应用于开源和商业项目中,它有助于简化编译过程,并支持生成多种开发环境下的构建配置。
CMake 3.25.3版本作为该系列软件包中的一个点,是CMake的一个稳定版本,它为开发者提供了一系列新特性和改进。随着版本的更新,3.25.3版本可能引入了新的命令、改进了用户界面、优化了构建效率或解决了之前版本中发现的问题。
CMake的主要特点包括:
1. 跨平台性:CMake支持多种操作系统和编译器,包括但不限于Windows、Linux、Mac OS、FreeBSD、Unix等。
2. 编译器独立性:CMake生成的构建文件与具体的编译器无关,允许开发者在不同的开发环境中使用同一套构建脚本。
3. 高度可扩展性:CMake能够使用CMake模块和脚本来扩展功能,社区提供了大量的模块以支持不同的构建需求。
4. CMakeLists.txt:这是CMake的配置脚本文件,用于指定项目源文件、库依赖、自定义指令等信息。
5. 集成开发环境(IDE)支持:CMake可以生成适用于多种IDE的项目文件,例如Visual Studio、Eclipse、Xcode等。
6. 命令行工具:CMake提供了命令行工具,允许用户通过命令行对构建过程进行控制。
7. 可配置构建选项:CMake支持构建选项的配置,使得用户可以根据需要启用或禁用特定功能。
8. 包管理器支持:CMake可以从包管理器中获取依赖,并且可以使用FetchContent或ExternalProject模块来获取外部项目。
9. 测试和覆盖工具:CMake支持添加和运行测试,并集成代码覆盖工具,帮助开发者对代码进行质量控制。
10. 文档和帮助系统:CMake提供了一个内置的帮助系统,可以为用户提供命令和变量的详细文档。
CMake的安装和使用通常分为几个步骤:
- 下载并解压对应平台的CMake软件包。
- 在系统中配置CMake的环境变量,确保在命令行中可以全局访问cmake命令。
- 根据项目需要编写CMakeLists.txt文件。
- 在含有CMakeLists.txt文件的目录下执行cmake命令生成构建文件。
- 使用生成的构建文件进行项目的构建和编译工作。
CMake的更新和迭代通常会带来更好的用户体验和更高效的构建过程。对于开发者而言,及时更新到最新稳定版本的CMake是保持开发效率和项目兼容性的重要步骤。而对于新用户,掌握CMake的使用则是学习现代软件构建技术的一个重要方面。"
"互动学习:行动中的多样性与论文攻读经历"
多样性她- 事实上SCI NCES你的时间表ECOLEDO C Tora SC和NCESPOUR l’Ingén学习互动,互动学习以行动为中心的强化学习学会互动,互动学习,以行动为中心的强化学习计算机科学博士论文于2021年9月28日在Villeneuve d'Asq公开支持马修·瑟林评审团主席法布里斯·勒菲弗尔阿维尼翁大学教授论文指导奥利维尔·皮耶昆谷歌研究教授:智囊团论文联合主任菲利普·普雷教授,大学。里尔/CRISTAL/因里亚报告员奥利维耶·西格德索邦大学报告员卢多维奇·德诺耶教授,Facebook /索邦大学审查员越南圣迈IMT Atlantic高级讲师邀请弗洛里安·斯特鲁布博士,Deepmind对于那些及时看到自己错误的人...3谢谢你首先,我要感谢我的两位博士生导师Olivier和Philippe。奥利维尔,"站在巨人的肩膀上"这句话对你来说完全有意义了。从科学上讲,你知道在这篇论文的(许多)错误中,你是我可以依
数字信号处理全攻略:掌握15个关键技巧,提升你的处理效率
# 摘要
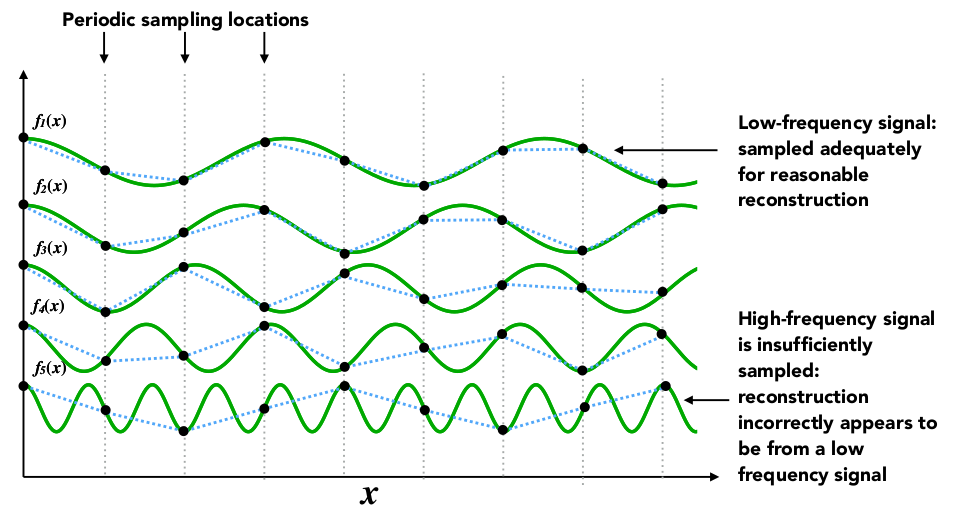
数字信号处理作为信息科技领域的重要分支,涵盖了从基础理论到高级应用的广泛内容。本文首先介绍了数字信号处理的基础知识,包括基本概念、时域与频域分析、以及核心算法和理论框架。接着,详细探讨了实现信号采样和量化、滤波器设计、以及信号增强和重建实践技巧。文章进一步深入到高级应用,包括信号压缩技术、多维信号处理,以及人工智能技术在信号处理中的集成。最后,本文评述了数字信号处理
给定不超过6的正整数A,考虑从A开始的连续4个数字。请输出所有由它们组成的无重复数字的3位数。编写一个C语言程序
为了编写一个C语言程序来解决这个问题,我们需要遍历给定范围内的所有连续4个数字,并检查每个组合是否能构成一个无重复数字的三位数。这里是一个简单的示例程序:
```c
#include <stdio.h>
// 函数定义,用于生成并检查无重复数字的3位数
void generate_unique_3_digit(int A) {
for (int i = A; i <= A + 3; i++) {
int num = i * 100 + (i+1) * 10 + (i+2);
if (num >= 100 && num < 1000 && is_uni
直流无刷电机控制技术项目源码集合
资源摘要信息:"直流无刷实例源码.zip"
该资源为一个包含多个技术项目源码的压缩文件,涵盖了IT技术的多个领域。接下来将详细介绍这些领域,并对其在源码中的应用进行说明。
1. 前端开发:前端开发通常指使用HTML、CSS和JavaScript等技术进行网页界面的构建。前端源码可能包括实现用户交互界面的代码,响应式布局实现,以及一些前端框架(如React或Vue.js)的使用实例。
2. 后端开发:后端通常涉及服务器端的编程,使用如PHP、Java、Python、C#等语言,处理HTTP请求、数据库交互、业务逻辑实现等。源码中可能包含服务器的搭建、数据库设计、API接口的实现等方面的内容。
3. 移动开发:移动开发关注于移动设备上的应用开发,涉及iOS、Android等平台,使用Swift、Kotlin、Java或跨平台框架如Flutter等。源码可能包括移动界面的布局、触摸事件处理、应用与后端数据的交互等。
4. 操作系统:操作系统源码可能包括对Linux内核的修改、或是基于RTOS(实时操作系统)的嵌入式系统开发。这类源码往往更偏向底层,涉及系统级编程。
5. 人工智能:人工智能项目源码可能包含机器学习、深度学习的实现,使用Python的TensorFlow或PyTorch框架等。这些源码可能涉及图像识别、自然语言处理等复杂算法的实现。
6. 物联网:物联网项目源码可能包含设备端与云平台的数据交互,使用的技术可能包括MQTT协议、HTTP/HTTPS协议等,可能还会涉及ESP8266这样的Wi-Fi模块使用。
7. 信息化管理:这类项目源码可能包含企业信息系统的构建,使用的技术可能包括数据库操作、数据报表生成、工作流管理等。
8. 数据库:数据库源码可能包括数据库的设计、操作,比如使用MySQL、PostgreSQL、MongoDB等数据库系统的SQL编写、存储过程、触发器等。
9. 硬件开发:硬件开发源码可能涉及使用STM32微控制器、EDA工具(如Proteus)进行电路设计、模拟和编程。
10. 大数据:大数据源码可能包含数据采集、存储、处理和分析的过程,可能会用到Hadoop、Spark、Flink等大数据处理框架。
11. 课程资源:这部分源码可能是为教学目的设计的,它可能包括一些基本项目的实现,适合初学者学习和理解。
12. 音视频:音视频源码可能包括音视频播放、录制、编解码等技术的应用,可能涉及到webRTC、FFmpeg等技术。
13. 网站开发:网站开发源码可能包括从简单的静态页面到复杂的动态网站实现,涉及前端框架、后端逻辑、数据库交互等。
14. EDA:电子设计自动化(EDA)源码可能包括电路图设计、PCB布线等,使用如Altium Designer、Eagle等专业EDA工具。
15. Proteus:Proteus源码可能包括电路的模拟和测试,它可以模拟微控制器和其他电子元件的行为。
该资源所包含的项目源码均已通过严格测试,可以直接运行。源码的适用人群广泛,不仅适合初学者学习不同技术领域,也适合进阶学习者或专业人士作为参考或直接拿来修改扩展,实现新功能。所有源码的上传都经过确认其正常工作,确保下载者可以直接使用。
在使用这些源码时,如果遇到任何问题,可以随时与博主沟通,博主将提供及时的解答。此外,鼓励用户下载和使用这些资源,互相学习、共同进步。
由于压缩文件的文件名称列表中只提供了"直流无刷实例源码",没有具体项目名称,因此我们无法得知具体的项目实例。然而,根据文件描述,我们可以确定这些源码项目覆盖了从硬件到软件、从传统应用到现代技术的广泛范围,并且针对了直流无刷电机的控制实例进行了特别的说明。
请注意,由于资源的宽泛涵盖性,这里提供的信息并不包含特定项目的详细分析,而是根据描述中的关键词进行了技术领域的概括性描述。如果需要针对具体项目进行分析,建议下载资源并根据具体文件内容进行详细探讨。