蛋白质结构预测与分子模拟方法
发布时间: 2024-03-21 17:52:38 阅读量: 30 订阅数: 17 
# 1. 蛋白质结构预测简介
## 1.1 什么是蛋白质结构预测
蛋白质结构预测是指通过计算机模拟和算法预测蛋白质的三维空间结构,包括螺旋、β折叠等二级结构以及整体的立体构象。它是生物信息学领域的重要研究方向之一,也是分子生物学和药物设计的基础。
## 1.2 蛋白质结构预测的意义与应用
蛋白质结构预测的准确性直接关系到后续对蛋白质功能、相互作用以及药物设计的研究和应用。通过预测蛋白质结构可以更好地理解蛋白质的生物学功能,加速药物研发过程,优化药物设计。
## 1.3 蛋白质结构预测的方法概述
蛋白质结构预测的方法主要包括生物信息学方法、分子模拟方法和实验方法。生物信息学方法包括序列比对、结构比对等;分子模拟方法包括分子动力学模拟、Monte Carlo 模拟等;实验方法则是通过X射线晶体学、核磁共振等技术来确定蛋白质结构。综合利用多种方法可以提高蛋白质结构预测的准确性和可靠性。
# 2. 蛋白质结构预测的生物信息学方法
在蛋白质结构预测领域,生物信息学方法是一种常用的手段,可以通过分析蛋白质序列及其相似性来推测蛋白质的结构。本章将介绍基于生物信息学方法的蛋白质结构预测技术。
### 2.1 基于序列相似性的结构预测方法
基于序列相似性的方法是一种常见的蛋白质结构预测技术,它通过比对目标蛋白质序列与已知蛋白质序列的相似性,来推测目标蛋白质的结构。该方法包括以下步骤:
```python
# 示例代码:基于序列相似性的结构预测方法
from Bio import pairwise2
from Bio.pairwise2 import format_alignment
# 目标蛋白质序列
target_protein = "MSEQET"
# 已知蛋白质序列
known_protein = "MQQET"
# 序列比对
alignments = pairwise2.align.globalxx(target_protein, known_protein)
for alignment in alignments:
print(format_alignment(*alignment))
```
**代码总结:** 以上代码演示了基于序列相似性的结构预测方法,通过比对目标蛋白质序列与已知蛋白质序列的相似性,输出两者的序列比对结果。
**结果说明:** 通过序列比对,可以找到目标蛋白质与已知蛋白质之间的相似性,进而推测目标蛋白质的结构。
### 2.2 结构比对与结构基因组学
结构比对和结构基因组学是生物信息学中重要的技术手段,可用于比对蛋白质结构、寻找结构保守域等。结构比对通常包括以下步骤:
```java
// 示例代码:结构比对
public class StructureAlignment {
public static void main(String[] args) {
// 读取两个蛋白质结构文件
Structure protein1 = readStructureFile("protein1.pdb");
Structure protein2 = readStructureFile("protein2.pdb");
// 进行结构比对
StructureAlignment algorithm = new StructureAlignment();
algorithm.alignStructures(protein1, protein2);
}
}
```
### 2.3 基于折叠动力学的结构预测算法
折叠动力学是一种基于蛋白质结构动力学性质的结构预测算法,通过模拟蛋白质结构的折叠过程来推测其最稳定的构象。以下是一个简单的基于折叠动力学的结构预测算法示例:
```go
// 示例代码:基于折叠动力学的结构预测算法
package main
import "fmt"
func main() {
// 模拟蛋白质折叠过程
proteinSequence := "MKLTIET"
foldedStructure := foldProtein(proteinSequence)
fmt.Println("Predic
```
 最低0.47元/天 解锁专栏
最低0.47元/天 解锁专栏 VIP年卡限时特惠
 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏深入探讨了生物信息学与基因组学领域的多个重要主题。从生物信息学与基因组学的导论到基因组学中的序列比对算法,再到基因组组装技术的原理与方法介绍,以及基因结构预测、蛋白质结构预测等多方面内容的讨论,涵盖了该领域的广泛知识。同时,也介绍了转录组测序技术、遗传变异检测、基因编辑技术等前沿技术以及其应用。此外,本专栏还关注了表观基因组学、进化基因组学、微生物组学等新兴研究领域,揭示了生物信息学在机器学习、深度学习等方面的应用,以及功能性基因组学与代谢组学的结合分析。通过全面而深入的内容,旨在帮助读者深入了解生物信息学与基因组学领域的研究进展与方法应用。
专栏目录
最低0.47元/天 解锁专栏
VIP年卡限时特惠
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
MATLAB求平均值在社会科学研究中的作用:理解平均值在社会科学数据分析中的意义

# 1. 平均值在社会科学中的作用
平均值是社会科学研究中广泛使用的一种统计指标,它可以提供数据集的中心趋势信息。在社会科学中,平均值通常用于描述人口特
深入了解MATLAB开根号的最新研究和应用:获取开根号领域的最新动态

# 1. MATLAB开根号的理论基础
开根号运算在数学和科学计算中无处不在。在MATLAB中,开根号可以通过多种函数实现,包括`sqrt()`和`nthroot()`。`sqrt()`函数用于计算正实数的平方根,而`nt
MATLAB符号数组:解析符号表达式,探索数学计算新维度

# 1. MATLAB 符号数组简介**
MATLAB 符号数组是一种强大的工具,用于处理符号表达式和执行符号计算。符号数组中的元素可以是符
MATLAB柱状图在信号处理中的应用:可视化信号特征和频谱分析
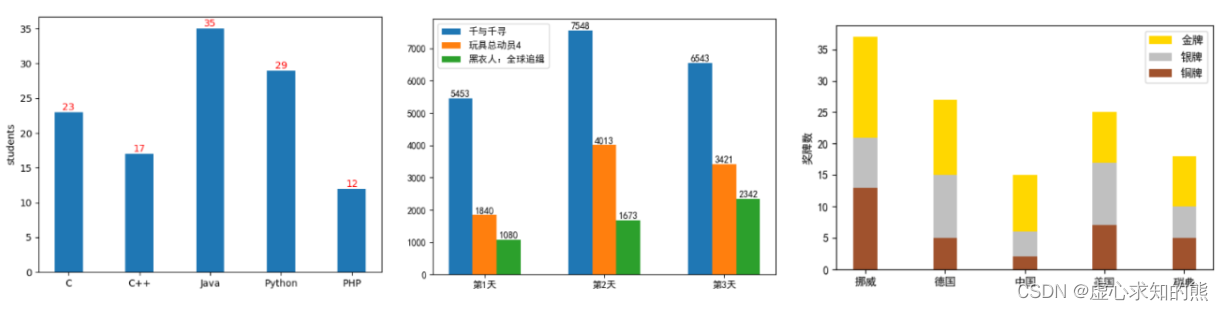
# 1. MATLAB柱状图概述**
MATLAB柱状图是一种图形化工具,用于可视化数据中不同类别或组的分布情况。它通过绘制垂直条形来表示每个类别或组中的数据值。柱状图在信号处理中广泛用于可视化信号特征和进行频谱分析。
柱状图的优点在于其简单易懂,能够直观地展示数据分布。在信号处理中,柱状图可以帮助工程师识别信号中的模式、趋势和异常情况,从而为信号分析和处理提供有价值的见解。
# 2. 柱状图在信号处理中的应用
柱状图在信号处理
NoSQL数据库实战:MongoDB、Redis、Cassandra深入剖析
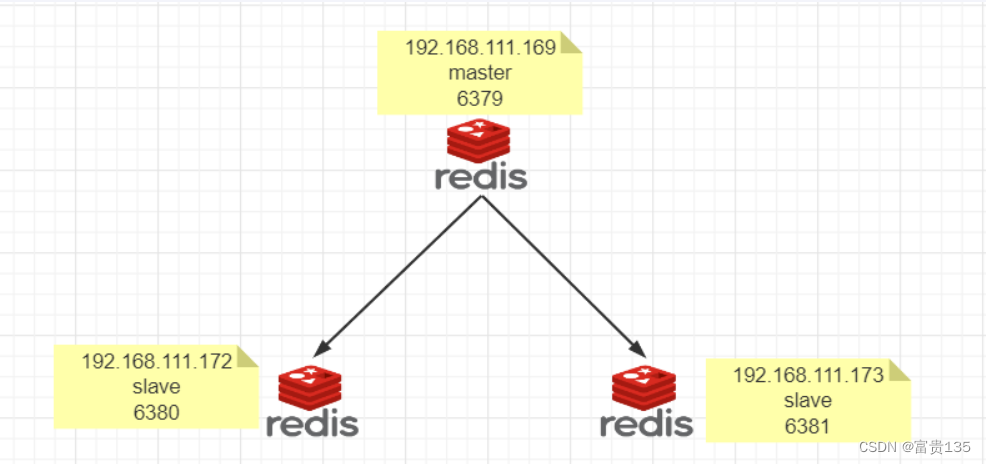
# 1. NoSQL数据库概述
**1.1 NoSQL数据库的定义**
NoSQL(Not Only SQL)数据库是一种非关系型数据库,它不遵循传统的SQL(结构化查询语言)范式。NoSQL数据库旨在处理大规模、非结构化或半结构化数据,并提供高可用性、可扩展性和灵活性。
**1.2 NoSQL数据库的类型**
NoSQL数据库根据其数据模型和存储方式分为以下
MATLAB在图像处理中的应用:图像增强、目标检测和人脸识别
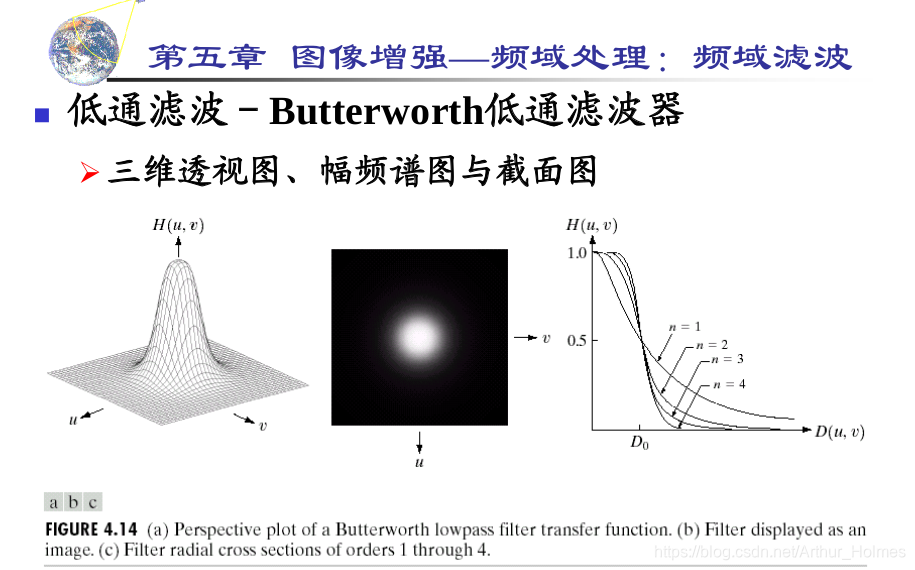
# 1. MATLAB图像处理概述
MATLAB是一个强大的技术计算平台,广泛应用于图像处理领域。它提供了一系列内置函数和工具箱,使工程师
MATLAB字符串拼接与财务建模:在财务建模中使用字符串拼接,提升分析效率

# 1. MATLAB 字符串拼接基础**
字符串拼接是 MATLAB 中一项基本操作,用于将多个字符串连接成一个字符串。它在财务建模中有着广泛的应用,例如财务数据的拼接、财务公式的表示以及财务建模的自动化。
MATLAB 中有几种字符串拼接方法,包括 `+` 运算符、`strcat` 函数和 `sprintf` 函数。`+` 运算符是最简单的拼接
MATLAB平方根硬件加速探索:提升计算性能,拓展算法应用领域

# 1. MATLAB 平方根计算基础**
MATLAB 提供了 `sqrt()` 函数用于计算平方根。该函数接受一个实数或复数作为输入,并返回其平方根。`sqrt()` 函数在 MATLAB 中广泛用于各种科学和工程应用中,例如信号处理、图像处理和数值计算。
**代码块:**
```matlab
% 计算实数的平方根
x = 4;
sqrt_x = sqrt(x);
%
MATLAB散点图:使用散点图进行信号处理的5个步骤
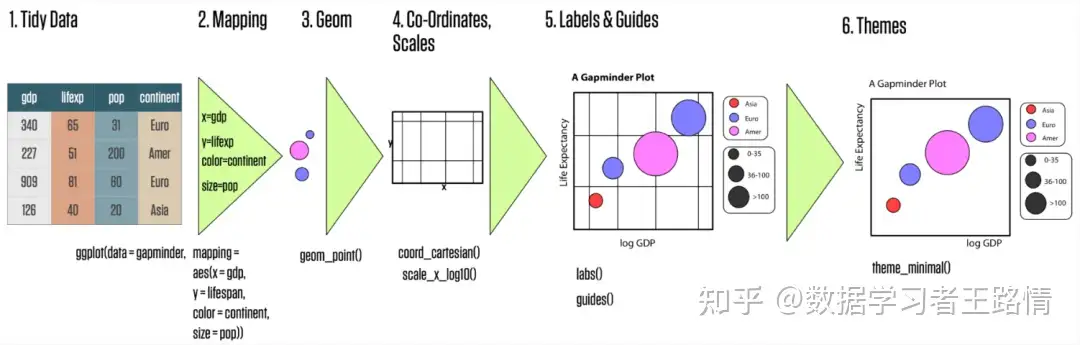
# 1. MATLAB散点图简介
散点图是一种用于可视化两个变量之间关系的图表。它由一系列数据点组成,每个数据点代表一个数据对(x,y)。散点图可以揭示数据中的模式和趋势,并帮助研究人员和分析师理解变量之间的关系。
在MATLAB中,可以使用`scatter`函数绘制散点图。`scatter`函数接受两个向量作为输入:x向量和y向量。这些向量必须具有相同长度,并且每个元素对(x,y)表示一个数据点。例如,以下代码绘制
图像处理中的求和妙用:探索MATLAB求和在图像处理中的应用

# 1. 图像处理简介**
图像处理是利用计算机对图像进行各种操作,以改善图像质量或提取有用信息的技术。图像处理在各个领域都有广泛的应用,例如医学成像、遥感、工业检测和计算机视觉。
图像由像素组成,每个像素都有一个值,表示该像素的颜色或亮度。图像处理操作通常涉及对这些像素值进行数学运算,以达到增强、分
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
VIP年卡限时特惠
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )