【R语言生态学数据分析】:vegan包使用指南,探索生态学数据的奥秘
发布时间: 2024-11-08 19:49:04 阅读量: 663 订阅数: 43 

数量生态学代码及R包
# 1. R语言在生态学数据分析中的应用
生态学数据分析的复杂性和多样性使其成为现代科学研究中的一个挑战。R语言作为一款免费的开源统计软件,因其强大的统计分析能力、广泛的社区支持和丰富的可视化工具,已经成为生态学研究者不可或缺的工具。在本章中,我们将初步探索R语言在生态学数据分析中的应用,从了解生态学数据的特点开始,过渡到掌握R语言的基础操作,最终将重点放在如何通过R语言高效地处理和解释生态学数据。我们将通过具体的例子和案例分析,展示R语言如何解决生态学中遇到的实际问题,帮助研究者更深入地理解生态系统的复杂性,从而做出更为精确和可靠的科学结论。
# 2. vegan包基础与理论框架
## 2.1 生态学数据的类型与特点
### 2.1.1 多样性指数与物种丰富度
生态学研究的核心之一是度量和分析物种的多样性和丰富度。多样性指数是评估生物多样性复杂性和变化的量化工具,而物种丰富度则反映了某地区物种数量的多少。在R语言中,vegan包提供了多种多样性指数的计算方法,包括但不限于Shannon、Simpson以及物种丰富度指数。
```r
# 安装并加载vegan包
install.packages("vegan")
library(vegan)
# 使用example数据集
data(BCI)
# 计算物种丰富度
richness <- specnumber(BCI)
# 计算Shannon多样性指数
shannon_diversity <- diversity(BCI)
```
在上述代码中,`specnumber`函数用于计算物种丰富度,而`diversity`函数则计算了Shannon多样性指数。`BCI`是vegan包中的一个示例数据集,代表了巴拿马岛的一个森林样方调查数据。
### 2.1.2 相似性与差异性度量
生态学中的物种相似性或差异性度量是通过各种统计方法来确定不同样方之间的物种组成相似程度。常用的度量方法包括Jaccard指数和Bray-Curtis指数。Jaccard指数关注的是样方间共有物种与总物种的比例,而Bray-Curtis指数则基于物种丰度信息,更加关注物种数量的差异。
```r
# 计算Bray-Curtis相似性指数
bray_curtis <- vegdist(BCI, method = "bray")
# 计算Jaccard相似性指数
jaccard_index <- vegdist(BCI, method = "jaccard")
```
在此代码块中,`vegdist`函数用于计算生态学数据中样方间的相似性或差异性指数。用户需要指定相似性指数的种类,这里通过`method`参数分别计算了Bray-Curtis和Jaccard指数。
## 2.2 vegan包的安装与初步使用
### 2.2.1 安装vegan包
R语言的包管理非常简单,但安装vegan包之前需要确保已经安装了R语言环境。使用下面的代码进行安装:
```r
install.packages("vegan")
```
安装vegan包后,我们就可以开始使用它的功能来进行生态学数据分析了。接着需要加载该包,使其功能在当前的R会话中可用:
```r
library(vegan)
```
### 2.2.2 基本函数与数据集探索
安装并加载vegan包后,我们来探索一些其提供的基本函数和数据集。vegan包包含了几个用于教学和示例的数据集,比如之前提到的`BCI`数据集。我们可以使用`data()`函数来查看所有可用的示例数据集:
```r
# 查看所有可用的示例数据集
data(package = "vegan")
```
使用`str()`函数可以帮助我们了解数据集的基本结构和变量类型:
```r
# 查看数据集BCI的结构
str(BCI)
```
## 2.3 理解多元统计方法在生态学中的应用
### 2.3.1 主成分分析(PCA)和对应分析(CA)
主成分分析(PCA)和对应分析(CA)都是用于揭示数据结构的降维技术。PCA通过正交变换将一组可能相关的变量转换为一组线性不相关的变量,这些新变量称为主成分。而CA则特别适用于分析多维交叉表的数据结构,适用于生态学中的物种丰度数据。
```r
# 对BCI数据集进行PCA分析
pca_result <- rda(BCI)
# 进行CA分析
ca_result <- cca(BCI ~ 1)
```
在这里,`rda`函数执行了PCA分析,而`cca`函数则执行了对应分析。在PCA分析中,`BCI`数据集被转换为一组主成分,可以用来在二维或三维空间中可视化样方间的相互关系。对应分析则适用于识别物种和样方之间的结构。
### 2.3.2 群落排序(NMDS)与聚类分析(CLUSTER)
群落排序(NMDS)和聚类分析(CLUSTER)是两种进一步探索群落结构的方法。NMDS是一种非线性方法,可以用来探索和可视化样本间的相似性或差异性。聚类分析则可以将样方根据物种组成相似性划分成不同的群落类型。
```r
# 对BCI数据集进行NMDS分析
nmds_result <- metaMDS(BCI)
# 进行聚类分析
cluster_result <- hclust(dist(BCI), method = "complete")
```
在上面的代码中,`metaMDS`函数执行了非度量多维尺度分析(NMDS),而`hclust`函数则基于Bray-Curtis距离矩阵执行了层次聚类分析。这两种分析方法均能揭示样本之间的群落结构关系。
```mermaid
graph TD;
A[开始分析] --> B[安装vegan包]
B --> C[加载vegan包]
C --> D[探索数据集]
D --> E[进行PCA分析]
D --> F[执行CA分析]
D --> G[执行NMDS分析]
D --> H[执行聚类分析]
```
通过上述的分析流程,生态学家可以深入探索物种组成的复杂关系,并理解不同物种群落之间的相互作用和分布模式。每一步都利用了vegan包的强大功能,展示了生态学数据分析的多样性和深度。
以上章节内容展示了vegan包在生态学数据分析中的基础与理论框架,接下来章节内容将继续深入到实践应用。通过真实数据集的分析,你将会学习如何在实际工作中应用这些理论知识。
# 3. vegan包的实践应用
在本章中,我们将深入探讨vegan包在实际生态学数据分析中的应用。通过这一章节,您将学会如何使用vegan包进行物种多样性分析、群落结构分析以及真实生态数据的案例研究。我们将从理论到实践,逐步揭示vegan包在生态学数据分析中的强大功能和应用技巧。
## 3.1 多样性分析实践
多样性分析是生态学研究的基础,涉及到物种丰富度和多样性的测定。vegan包提供了一系列用于计算和分析多样性的函数,能够帮助研究者深入理解群落的结构和功能。
### 3.1.1 物种丰富度与多样性指数的计算
物种丰富度是衡量群落中物种数量的指标,而多样性指数则结合了物种丰富度和均匀度,提供了更为全面的群落多样性度量。vegan包中的`specnumber`函数可以用来计算物种丰富度,而`diversity`函数则计算了诸如Shannon和Simpson等多样性指数。
```r
# 计算物种丰富度
library(vegan)
data(dune)
species_richness <- specnumber(dune)
# 计算Shannon多样性指数
shannon_index <- diversity(dune, index = "shannon")
```
在上面的代码中,`dune`数据集是vegan包自带的一个生态学数据集。`specnumber`函数用于计算数据集中每个样方的物种丰富度,结果存储在`species_richness`变量中。`diversity`函数则计算Shannon多样性指数,其`index`参数指定计算的多样性指数类型。
### 3.1.2 α-多样性、β-多样性与γ-多样性分析
多样性分析不仅仅是对单个样方的度量,还包括群落间的比较(β-多样性)以及大区域内的整体多样性(γ-多样性)。vegan包通过一系列函数支持这类多样性分析。
```r
# 计算β-多样性
beta_diversity <- betadisper(dune_env, dune)
# 计算γ-多样性
gamma_diversity <- diversity(rowSums(dune), index = "shannon")
```
在上面的代码示例中,`betadisper`函数用于计算β-多样性,它评估了群落的分散度。参数`dune_env`是群落环境数据,`dune`是群落物种数据。`gamma_diversity`的计算则是通过将所有样方的物种丰富度相加,并应用Shannon多样性指数得出。
## 3.2 生态学群落数据的分析
群落数据的分析对于理解生态学过程至关重要。vegan包提供了一系列工具,帮助我们可视化群落结构,以及探索物种与环境变量之间的关联。
### 3.2.1 群落结构的可视化
在生态学研究中,群落结构的可视化是理解数据的关键。vegan包中的`plot`函数结合`decorana`函数可以对群落进行二维排序,并生成排序图。
```r
# 进行群落排序
dune排序 <- decorana(dune)
# 可视化群落结构
plot(dune排序, type = "t")
```
在上述代码中,`decorana`函数根据物种组成对样方进行排序,结果存储在`dune排序`变量中。然后,使用`plot`函数绘制排序图,`type`参数指定绘图类型为“t”,代表文本标注的排序图。
### 3.2.2 物种与环境变量之间的关联分析
了解物种与环境变量之间的关系对于揭示生态学过程具有重要意义。vegan包中的`envfit`函数可以用于检验环境变量与群落排序结果之间的关系。
```r
# 环境拟合分析
fit <- envfit(dune排序, dune_env)
# 查看拟合结果
print(fit)
```
在该代码

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏提供了一系列详细的教程,涵盖了 R 语言中广泛使用的数据包。从数据处理和可视化到图论、时间序列分析、代码加速、并行计算和交互式应用开发,再到数据库操作、数据清洗、转换和地理空间数据处理,以及深度学习、贝叶斯统计和生态学数据分析,该专栏涵盖了 R 语言数据科学和统计分析的各个方面。通过这些教程,读者可以深入了解每个数据包的功能和使用方法,从而提升他们的 R 语言技能并有效地处理和分析数据。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
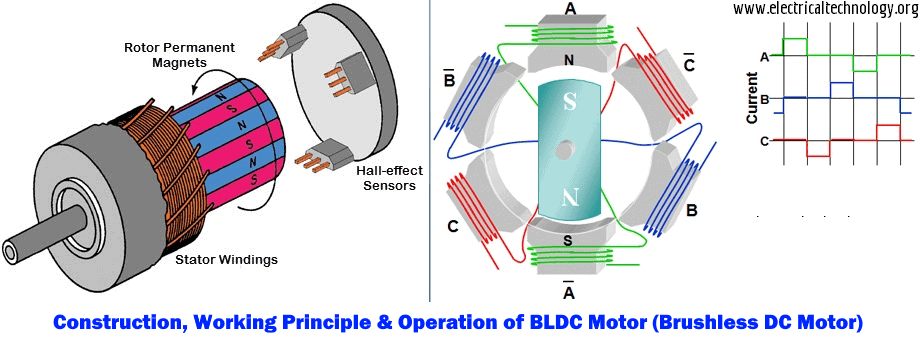
JY01A直流无刷IC全攻略:深入理解与高效应用
# 摘要
本文详细介绍了JY01A直流无刷IC的设计、功能和应用。文章首先概述了直流无刷电机的工作原理及其关键参数,随后探讨了JY01A IC的功能特点以及与电机集成的应用。在实践操作方面,本文讲解了JY01A IC的硬件连接、编程控制,并通过具体
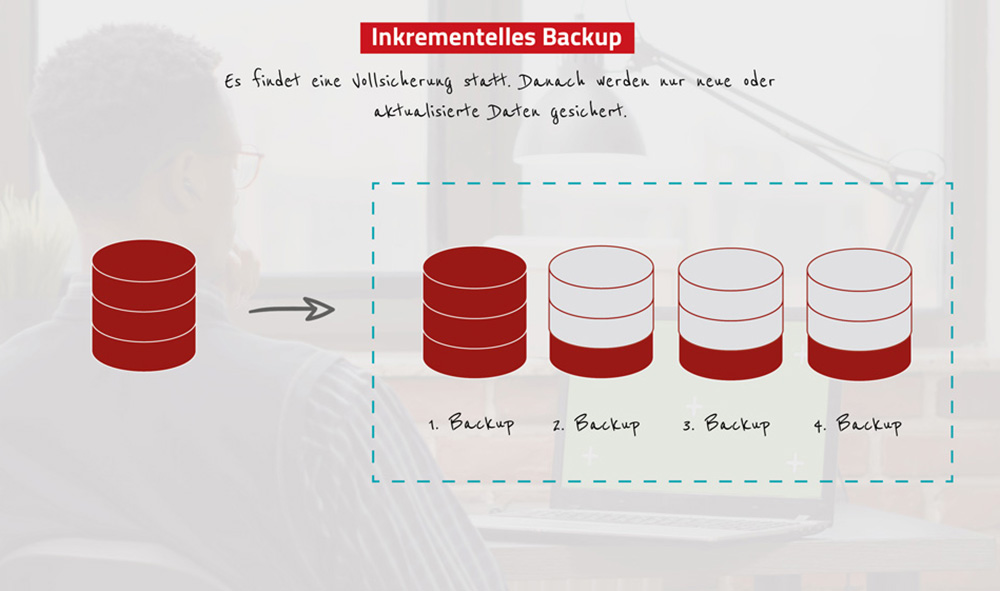
数据备份与恢复:中控BS架构考勤系统的策略与实施指南
# 摘要
在数字化时代,数据备份与恢复已成为保障企业信息系统稳定运行的重要组成部分。本文从理论基础和实践操作两个方面对中控BS架构考勤系统的数据备份与恢复进行深入探讨。文中首先阐述了数据备份的必要性及其对业务连续性的影响,进而详细介绍了不同备份类型的选择和备份周期的制定。随后,文章深入解析了数据恢复的原理与流程,并通过具体案例分析展示了恢复技术的实际应用。接着,本文探讨
【TongWeb7负载均衡秘笈】:确保请求高效分发的策略与实施
.webp)
# 摘要
本文从基础概念出发,对负载均衡进行了全面的分析和阐述。首先介绍了负载均衡的基本原理,然后详细探讨了不同的负载均衡策略及其算法,包括轮询、加权轮询、最少连接、加权最少连接、响应时间和动态调度算法。接着,文章着重解析了TongWeb7负载均衡技术的架构、安装配置、高级特性和应用案例。在实施案例部分,分析了高并发Web服务和云服务环境下负载
【Delphi性能调优】:加速进度条响应速度的10项策略分析

# 摘要
本论文首先概述了信号定位技术的基本概念和重要性,随后深入分析了三角测量和指纹定位两种主要技术的工作原理、实际应用以及各自的优势与不足。通过对三角测量定位模型的解析,我们了解到其理论基础、精度影响因素以及算法优化策略。指纹定位技术部分,则侧重于其理论框架、实际操作方法和应用场
【PID调试实战】:现场调校专家教你如何做到精准控制

# 摘要
PID控制作为一种历史悠久的控制理论,一直广泛应用于工业自动化领域中。本文从基础理论讲起,详细分析了PID参数的理论分析与选择、调试实践技巧,并探讨了PID控制在多变量、模糊逻辑以及网络化和智能化方面的高级应用。通过案例分析,文章展示了PID控制在实际工业环境中的应用效果以及特殊环境下参数调整的策略。文章最后展望了PID控制技术的发展方
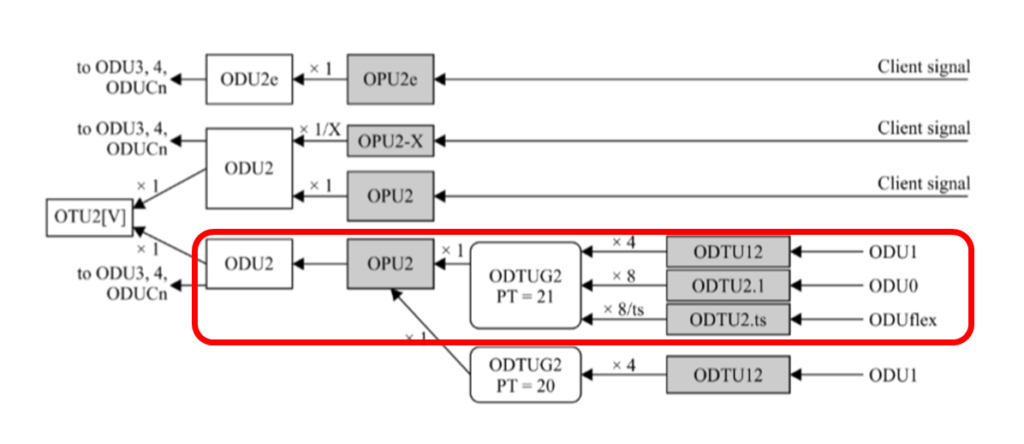
网络同步新境界:掌握G.7044标准中的ODU flex同步技术
# 摘要
本文详细探讨了G.7044标准与ODU flex同步技术,首先介绍了该标准的技术原理,包括时钟同步的基础知识、G.7044标准框架及其起源与应用背景,以及ODU flex技术
字符串插入操作实战:insert函数的编写与优化
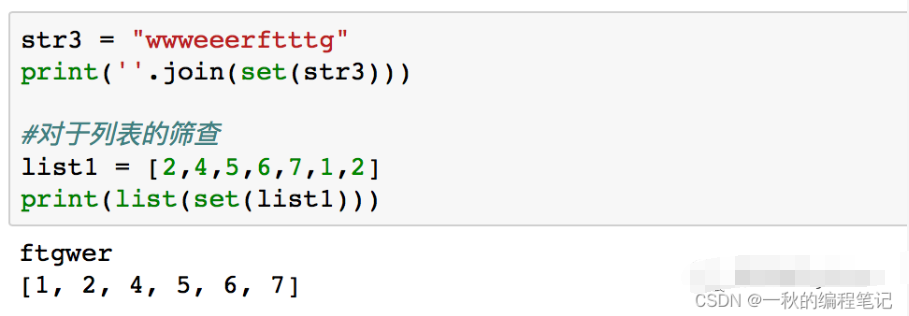
# 摘要
字符串插入操作是编程中常见且基础的任务,其效率直接影响程序的性能和可维护性。本文系统地探讨了字符串插入操作的理论基础、insert函数的编写原理、使用实践以及性能优化。首先,概述了insert函数的基本结构、关键算法和代码实现。接着,分析了在不同编程语言中insert函数的应用实践,并通过性能测试揭示了各种实现的差异。此外,本文还探讨了性能优化策略,包括内存使用和CPU效率提升,并介绍了高级数据结
环形菜单的兼容性处理

# 摘要
环形菜单作为一种用户界面元素,为软件和网页设计提供了新的交互体验。本文首先介绍了环形菜单的基本知识和设计理念,重点探讨了其通过HTML、CSS和JavaScript技术实现的方法和原理。然后,针对浏览器兼容性问题,提出了有效的解决方案,并讨论了如何通过测试和优化提升环形菜单的性能和用户体验。本
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )