MATLAB与化学工具箱:化学计算与建模的强大工具
发布时间: 2024-05-25 08:45:59 阅读量: 152 订阅数: 46 

matlab非常有用的工具箱

# 1. MATLAB简介**
MATLAB(Matrix Laboratory)是一种用于数值计算、数据分析和可视化的强大编程语言和环境。它因其在工程、科学和金融等领域的广泛应用而闻名。MATLAB提供了广泛的工具和函数,使其成为解决复杂计算问题的理想选择。
MATLAB使用矩阵为基础的数据结构,这使得它特别适合处理大型数据集和进行数值计算。它还具有一个交互式开发环境,允许用户快速原型化和调试代码。此外,MATLAB拥有一个庞大的用户社区和广泛的文档,为用户提供了丰富的支持和资源。
# 2. MATLAB化学工具箱概览
MATLAB化学工具箱是一个强大的工具包,为化学家和材料科学家提供了广泛的功能,用于分子建模、化学反应模拟、数据分析和可视化。它包含以下主要模块:
### 2.1 化学工具箱功能模块
#### 2.1.1 分子结构和性质计算
* **分子几何优化:**确定分子的最低能构型,用于预测键长、键角和二面角。
* **分子能级计算:**计算分子的电子能级和波函数,用于了解分子的电子结构和光谱性质。
* **分子性质计算:**计算分子极化率、偶极矩、表面积等物理性质。
#### 2.1.2 化学反应模拟
* **反应路径分析:**确定反应的过渡态和反应路径,用于理解反应机理。
* **反应速率常数计算:**计算反应速率常数,用于预测反应速率和反应产率。
* **化学动力学模拟:**模拟化学反应的动力学行为,用于研究反应机理和反应条件的影响。
#### 2.1.3 数据分析和可视化
* **数据拟合和回归:**对实验数据进行拟合和回归,用于提取参数和建立模型。
* **分子轨道可视化:**可视化分子的分子轨道,用于理解分子的电子分布和键合性质。
* **数据可视化:**创建各种类型的图表和图形,用于展示和分析数据。
### 2.2 化学工具箱使用入门
#### 2.2.1 工具箱安装和加载
* 下载并安装MATLAB化学工具箱。
* 在MATLAB命令窗口中,使用以下命令加载工具箱:
```
>> toolboxdir = fullfile(matlabroot, 'toolbox', 'chemtoolbox');
>> addpath(toolboxdir);
```
#### 2.2.2 基本命令和函数
* **创建分子结构:**使用`mol`函数创建分子结构对象,指定原子类型、键连接和坐标。
* **计算分子性质:**使用`getprop`函数计算分子的几何、能级和性质。
* **模拟化学反应:**使用`reaction`函数模拟化学反应,指定反应物、产物和反应条件。
* **数据分析和可视化:**使用`fit`函数进行数据拟合,使用`plot`函数创建图表和图形。
# 3. MATLAB化学工具箱实践应用
### 3.1 分子结构计算
分子结构计算是化学工具箱的重要功能之一,它可以帮助用户预测和分析分子的几何结构、能级和性质。
#### 3.1.1 分子几何优化
分子几何优化是确定分子最低能量构型的过程。MATLAB化学工具箱提供了多种方法来执行几何优化,包括:
- **Hartree-Fock (HF) 方法:**一种自洽场方法,它使用斯莱特行列式来近似分子波函数。
- **密度泛函理论 (DFT) 方法:**一种基于电子密度的第一性原理方法,它使用泛函来近似交换关联能。
- **分子力学 (MM) 方法:**一种基于经典力场的半经验方法,它使用势能函数来描述分子间的相互作用。
**代码块:**
```
% 使用 HF 方法优化水分子几何结构
molecule = 'H2O';
geometry = optimizeGeometry(molecule, 'Method', 'Hartree-Fock');
% 打印优化后的几何结构
disp(geometry);
```
**逻辑分析:**
* `optimizeGeometry` 函数用于执行几何优化。
* `molecule` 参数指定要优化的分子。
* `Method` 参数指定优化方法,在本例中为 Hartree-Fock 方法。
* `geometry` 变量存储优化后的几何结构。
* `disp` 函数用于打印几何结构。
#### 3.1.2 分子能级计算
分子能级计算是确定分子电子能级的过程。MATLAB化学工具箱提供了多种方法来计算分子能级,包括:
- **Hartree-Fock (HF) 方法:**一种自洽场方法,它使用斯莱特行列式来近似分子波函数。
- **密度泛函理论 (DFT) 方法:**一种基于电子密度的第一性原理方法,它使用泛函来近似交换关联能。
- **组态相互作用 (CI) 方法:**一种后 Hartree-Fock 方法,它考虑了电子相关性。
**代码块:**
```
% 使用 DFT 方法计算乙烯分子的能级
molecule = 'C2H4';
energy = calculateEnergy(molecule, 'Method', 'DFT');
% 打印计算出的能级
disp(energy);
```
**逻辑分析:**
* `calculateEnergy` 函数用于计算分子能级。
* `molecule` 参数指定要计算能级的分子。
* `Method` 参数指定计算方法,在本例中为 DFT 方法。
* `energy` 变量存储计算出的能级。
* `disp` 函数

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
《MATLAB不等于》专栏深入探讨了MATLAB与其他编程语言和工具箱之间的差异和互补性。文章涵盖了广泛的主题,包括:
* MATLAB与R在统计建模和数据挖掘中的比较
* MATLAB与C++在性能和效率方面的较量
* MATLAB与Java在跨平台开发和企业应用中的抉择
* MATLAB与Fortran在科学计算和高性能计算中的比拼
* MATLAB与App Designer在图形用户界面设计中的优势
* MATLAB与各种工具箱的集成,涵盖图像处理、信号分析、控制系统设计、优化、数据科学、机器学习、并行计算、云计算、物联网、生物信息学和化学计算等领域。
通过深入分析这些比较,该专栏旨在帮助读者了解MATLAB的独特优势和局限性,并为他们选择最适合其特定需求的工具提供指导。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【西数硬盘维修WDR5.3新手指南】:一步步教你基础入门和工具使用

# 摘要
本文系统介绍了西数硬盘维修软件WDR5.3的操作流程和技巧。文章首先概述了硬盘的工作原理和常见故障类型,随后详细阐释了WDR5.3软件的基本理论知识、操作实践、进阶技巧以及性能优化方法。通过详细分析真实案例,本文评估了维修前后的硬盘性能和数据恢复成功率。最后,文章总结了维修过程中的成功和失败经验,并对硬盘维修行业未来的发展趋势进行了展望。
# 关键字
硬盘维修;WDR5.3软件;故障诊断;数据恢复;性能
编程传奇:雷军如何用汇编代码重塑编程世界

# 摘要
本文全面探讨了汇编语言编程的历史演变、基础理论、编程实践技巧、雷军与汇编语言的关联故事以及其现代应用和未来展望。文章第一章回顾了汇编语言的发展历程
【BSF服务部署策略】:从理论到实际的转变

# 摘要
BSF服务部署策略是一个关键领域,涉及服务的概念、优势、部署环境、配置、优化和故障处理。本文全面概述了BSF服务的部署策略,提供了基础理论知识,并介绍了配置和优化的实际方法。文中还探讨了BSF服务的安全策略、集群部署和API集成
【智能电网新纪元】:继电保护技术的革新与IT融合
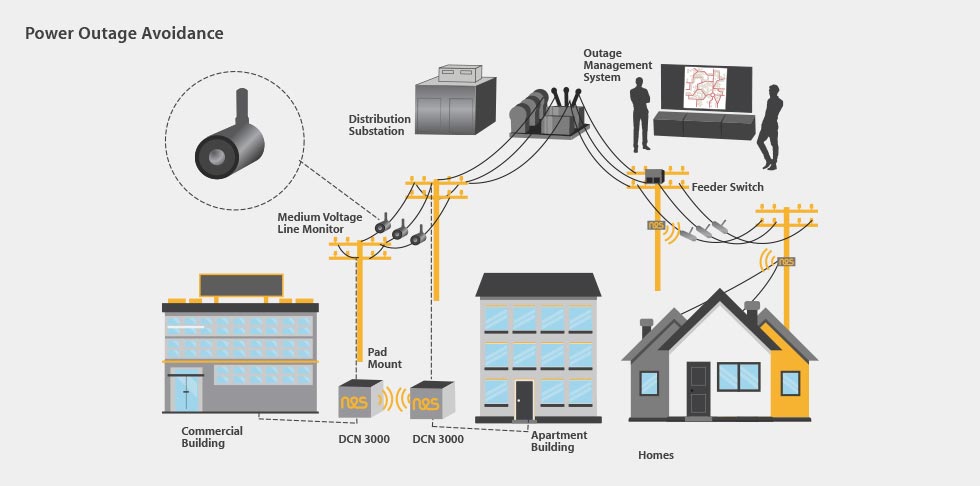
# 摘要
智能电网与继电保护技术是电力系统现代化的两大核心领域。本文首先概述了智能电网与继电保护技术的基本概念和理论基础,随后探讨了继电保护技术的创新进展和可靠性分析,同时分析了IT技术在继电保护领域的应用以及智能化系统架构和网络安全策略。在智能电网的IT技术融合实践章节,文章讨论了通信协议标准、IT系统实践案例和可持续发展策略。最后,文章展望了未来电网技术的发展方向,电网智能化面临的挑战和对策,并提出了创新与实践
【GMDSS通信原理揭秘】:深入理解与模拟实践技巧
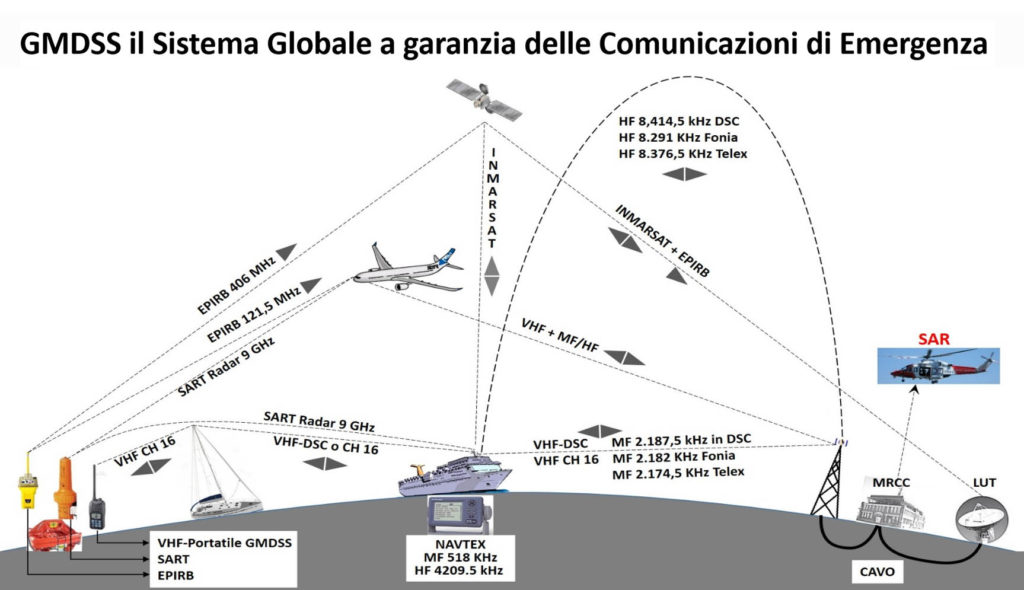
# 摘要
本文综述了全球海上遇险与安全系统(GMDSS)的通信技术,覆盖了硬件构成、通信协议、信号处理、模拟仿真,以及系统的安全与可靠性分析。在硬件构成方面,详细探讨了GMDSS主要设备的功能与分类、通信终端技术,以及导航设备与辅助系统。通信协议与信号部分介绍了GMDSS的标准协议、信号编码与调制技术,以及安全与紧急通信流程。模拟与仿真是通过软件进行通信测试和场景模拟,重点在于实验结果的分析与验证。安全与可靠性
【硬盘克隆进阶】:深入理解扇区级复制,个性化Ghost设置详解

# 摘要
随着信息技术的飞速发展,硬盘克隆技术已成为数据备份、迁移与恢复的重要手段。本文首先概述了硬盘克隆的基本概念及其在数据保护中的作用。随后,深入分析了扇区级复制的理论基础,包括硬盘结构、扇区定义及其复制原理。在个性化Ghost设置部分,本文详细介绍了Ghost软件的操作方法、硬件加速技巧以及扇区映射和错误检测的技术。通过实践操作部分,本文指导读者如何手动和通过自
FT232H接口设计:硬件与软件的考量要点

# 摘要
FT232H作为一种常用的USB转串口芯片,在数据通信领域发挥着重要作用。本文首先概述了FT232H接口的基本概念及其工作原理,然后深入分析了硬件设计的关键考量,包括电气特性、电源管理、PCB设计等。接着,文章探讨了软件驱动开发中固件与驱动架构、跨平台兼容性以及高级通信协议实现的重要性。通过不同领域应用实例的分析,展示了F
研发部门绩效考核案例研究:构建高效研发团队的KPI系统秘籍

# 摘要
绩效考核在研发团队管理中扮演着至关重要的角色,它直接关联到团队的工作效率和目标达成。本文深入探讨了KPI(关键绩效指标)与研发团队绩效之间的紧密联系,以及如何设计有效的KPI体系以确保其与组织目标的一致性。文章通过具体实践案例,分析了建立高效研发团队KPI系统的过程,并指出
【网络启动故障不求人】:一步步教你排查与解决PXE和GHOST常见问题

# 摘要
网络启动技术是现代IT基础设施部署中不可或缺的一部分,本文旨在探讨网络启动技术的基础原理、故障排查以及高级应用。首先,介绍了PXE启动技术及其故障排查,包括PXE的工作原理、常见故障类型和排查方法。接着,深入分析了GHOST部署中遇到的故障问题及其解决策略。此外,本文还探讨了网络启动的高级应用,例如集中管理和自动化部署,以及如何通过工具
STM32定时器高级应用:HAL库定时技巧与案例分析
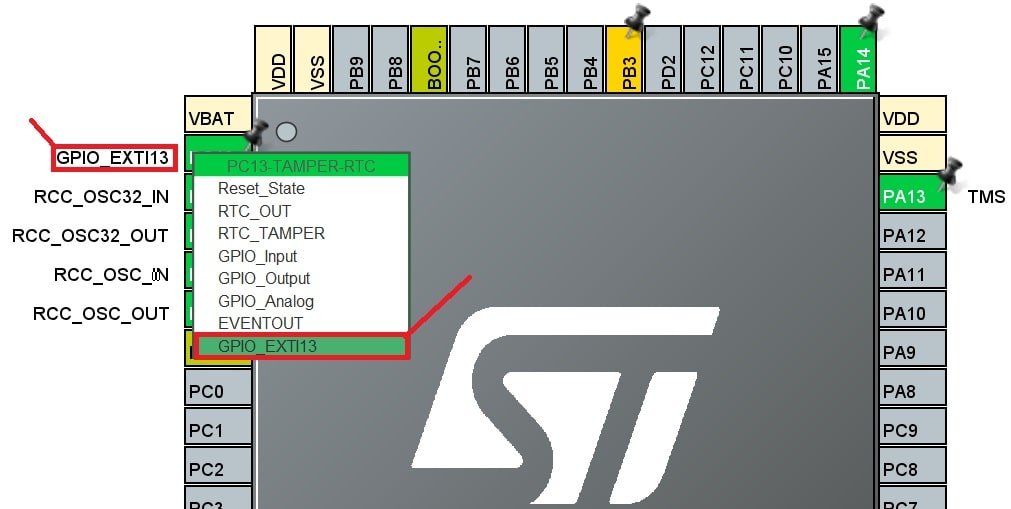
# 摘要
本文系统地探讨了STM32微控制器中定时器的功能、配置和应用。首先,介绍了定时器的基本工作原理和HAL库提供的API函数,以及定时器配置参数的详细解析。随后,本文深入阐述了定时器编程技巧,包括如何精确配置定时器时间和实现高级应用。文章进一步分析了定时器在不同应用场景中的实际运用,比如通信
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )