【DFT计算:从基础到进阶】
发布时间: 2024-12-06 11:05:37 阅读量: 22 订阅数: 16 

dft.rar_C语言DFT_DFT C_DFT C语言_dft c++_离散 DFT

参考资源链接:[Avogadro中文教程:分子建模与可视化全面指南](https://wenku.csdn.net/doc/6b8oycfkbf?spm=1055.2635.3001.10343)
# 1. DFT计算概述
计算化学和材料科学领域中,密度泛函理论(Density Functional Theory,DFT)是一种使用量子力学来解决多电子体系问题的方法。DFT计算方法因其相对较低的计算成本和较高的计算精度,已经成为了研究电子结构和材料性质的重要工具。
在本章中,我们将从应用、计算流程、以及理论基础三个方面简要介绍DFT计算。首先,概述DFT在现代科学领域中的广泛应用,包括材料设计、电池性能预测、表面反应研究等。然后,简述DFT计算的一般步骤,包括构建模型、优化结构、计算电子和光学性质等。最后,为后续章节铺垫,概括介绍密度泛函理论的基本概念及其在解决量子多体问题中的数学框架。通过这一章,读者将对DFT计算有一个总体的认识,并理解其在各领域的应用前景和潜力。
# 2. DFT基础理论
### 2.1 量子力学与固体物理基础
#### 2.1.1 量子力学的基本原理
量子力学作为描述微观粒子行为的理论框架,其基本原理奠定了现代物理和化学的基础。波函数是量子力学中的核心概念之一,它描述了微观粒子的量子态,并且随时间演化。波函数的平方绝对值表示粒子出现在某位置的概率密度。薛定谔方程是另一个基石,它是一个波动方程,描述了波函数随时间的演化。
理解量子力学原理对于掌握DFT至关重要,因为DFT是在量子力学框架内发展起来的。波函数的精确解往往需要复杂的数学计算,而DFT提供了计算固体中电子性质的替代方法,它通过计算粒子密度来简化问题,从而避免了直接处理波函数的复杂性。
#### 2.1.2 固体物理中的电子结构
固体物理关注的是晶体结构和电子行为。固体中的电子结构不仅决定了材料的电学性质,还影响了热学、光学和力学性质。在固体物理中,费米能级是一个非常重要的概念,它表示电子占据的能量状态分布的界限。
固体中的电子在周期性势场中运动,周期性势场来源于晶体中原子核的排列。电子的状态可以通过布洛赫波函数来描述,布洛赫波函数表征了电子在晶体格点上的周期性运动特性。电子结构的理论计算通常基于近似处理,而DFT利用电子密度作为基本变量,将多电子问题简化为单电子问题,提供了一种高效的计算手段。
### 2.2 密度泛函理论的基本概念
#### 2.2.1 Hohenberg-Kohn定理
Hohenberg-Kohn定理是密度泛函理论的基石,它包括了两个重要结论。第一个定理说明了多体薛定谔方程的基态物理性质可以由基态电子密度唯一决定。第二个定理证明了基态电子密度的泛函形式存在,并且其最小值对应于系统的基态能量。
这两个定理确立了电子密度作为描述系统基态属性的核心变量的地位,并且为后续DFT的发展提供了理论基础。Hohenberg-Kohn定理极大地简化了量子多体问题的复杂性,从而使得直接计算电子密度成为可能,而无需直接处理复杂的多电子波函数。
#### 2.2.2 Kohn-Sham方程
在Hohenberg-Kohn定理的基础上,Kohn-Sham提出了一个单电子模型,即所谓的Kohn-Sham方程,用来计算电子密度。在这个模型中,电子被假设为在非相互作用的势场中独立运动,相互作用效应用交换相关泛函来描述。
Kohn-Sham方程的形式上类似于经典的薛定谔方程,其中交换相关泛函是未知的,需要通过近似方法来确定。Kohn-Sham方程的成功在于它将复杂的多电子问题转化为一系列单电子问题的求解,极大地简化了计算过程,这为DFT的广泛应用提供了可能性。
### 2.3 DFT计算中的近似方法
#### 2.3.1 局域密度近似(LDA)
局域密度近似(LDA)是最简单的DFT近似方法之一,它假设系统中的电子密度在空间中是均匀的。在这种近似下,交换相关泛函只依赖于局部的电子密度。LDA在描述均匀电子气模型时非常准确,但对于实际的固体材料,这种近似可能会带来一定的误差。
尽管LDA存在一定的局限性,但由于其计算上的简便,它在实际DFT计算中仍然被广泛使用。LDA特别适合计算均匀或近均匀的体系,例如简单金属和半导体。LDA的成功在于它的有效性和计算效率,它为更复杂的近似方法奠定了基础。
#### 2.3.2 广义梯度近似(GGA)
广义梯度近似(GGA)是相对于LDA的改进,它不仅考虑了电子密度,还考虑了电子密度的梯度信息。在GGA中,交换相关泛函不仅依赖于电子密度,还依赖于电子密度的空间变化率。
GGA的引入显著提高了DFT计算在描述固体材料时的精度,特别是在处理电子密度不均匀的体系时,比如在固体界面、表面以及分子吸附等问题中。GGA能够更准确地描述电子间的交换相关作用,从而提供了比LDA更为精确的电子结构预测。
#### 2.3.3 杂化泛函和其它高级近似
为了进一步提高计算精度,杂化泛函被引入到DFT计算中。杂化泛函是将Hartree-Fock方法中的交换作用与DFT中的相关泛函相结合的一种方法。这种方法能够更精确地描述电子间的交换作用,从而提高了计算结果的准确性。
杂化泛函在处理某些特定类型的化学键和体系时表现得更为优越。然而,由于Hartree-Fock方法本身的计算代价较高,杂化泛函的计算成本通常高于传统的LDA和GGA方法。尽管如此,杂化泛函仍被广泛应用于需要高精度的计算场合。
此外,还有许多其他高级近似方法,如meta-GGA和自旋泛函等。这些方法在特定情况下能够提供更为详尽的信息,但同时它们也带来了更高的计算难度和复杂性。
```markdown
| 近似方法 | 描述 | 优点 | 缺点 |
| --- | --- | --- | --- |
| LDA | 局域密度近似,假设电子密度均匀 | 计算简单,效率高 | 对非均匀电子密度体系描述不准确 |
| GGA | 广义梯度近似,考虑电子密度的梯度 | 精度高于LDA,适用于多种体系 | 对某些体系仍可能不准确 |
| 杂化泛函 | 结合Hartree-Fock的交换项和DFT相关泛函 | 提高对电子交换作用的描述精度 | 计算成本高 |
```
在选择适当的DFT近似方法时,需要根据研究的具体体系和目标精度进行权衡。从简单的LDA到计算成本较高的杂化泛函,各种方法各有千秋,正确选择和应用近似方法对于获得准确的计算结果至关重要。
# 3. DFT计算工具与实践
DFT(Density Functional Theory)计算是一类强大的计算工具,它依赖于特定的软件来进行模拟计算。本章将深入探讨如何选择和安装DFT软件,以及如何进行DFT计算的基本步骤和高级操作。
## 3.1 DFT软件的选择与安装
### 3.1.1 常见DFT软件介绍
DFT计算的软件有许多种,每种软件都有其特定的功能和优缺点。以下是一些常用的DFT计算软件:
- **VASP (Vienna Ab-initio Simulation Package)**: 是一款非常流行的DFT计算软件,广泛应用于固体物理、材料科学和化学领域。VASP软件对固体材料的电子结构、原子结构、分子动力学等方面的研究提供了强大的计算支持。
- **QuantumESPRESSO**: 是一款开源的DFT计算软件包,它基于平面波和赝势技术,适用于复杂材料系统的计算。
- **Gaussian**: 主要用于量子化学计算,通过DFT方法可以模拟分子和反应过程。
- **ORCA**: 一款广泛使用的量子化学计算软件,它支持多种计算方法,包括DFT计算。
### 3.1.2 软

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【QT基础入门】:QWidgets教程,一步一个脚印带你上手
# 摘要
本文全面介绍了Qt框架的安装配置、Widgets基础、界面设计及进阶功能,并通过一个综合实战项目展示了这些知识点的应用。首先,文章提供了对Qt框架及其安装配置的简要介绍。接着,深入探讨了Qt Widgets,包括其基本概念、信号与槽机制、布局管理器等,为读者打下了扎实的Qt界面开发基础。文章进一步阐述了Widgets在界面设计中的高级用法,如标准控件的深入使用、资源文件和样式表的应用、界面国际化处理。进阶功能章节揭示了Qt对话框、多文档界面、模型/视图架构以及自定义控件与绘图的强大功能。最后,实战项目部分通过需求分析、问题解决和项目实现,展示了如何将所学知识应用于实际开发中,包括项目
数学魔法的揭秘:深度剖析【深入理解FFT算法】的关键技术

# 摘要
快速傅里叶变换(FFT)是信号处理领域中一项关键的数学算法,它显著地降低了离散傅里叶变换(DFT)的计算复杂度。本文从FFT算法的理论基础、实现细节、在信号处理中的应用以及编程实践等多方面进行了详细讨论。重点介绍了FFT算法的数学原理、复杂度分析、频率域特性,以及常用FFT变体和优化技术。同时,本文探讨了FFT在频谱分析、数字滤波器设计、声音和图像处理中的实
MTK-ATA技术入门必读指南:从零开始掌握基础知识与专业术语

# 摘要
MTK-ATA技术作为一种先进的通信与存储技术,已经在多个领域得到广泛应用。本文首先介绍了MTK-ATA技术的概述和基础理论,阐述了其原理、发展以及专业术语。随后,本文深入探讨了MTK-ATA技术在通信与数据存储方面的实践应用,分析了其在手机通信、网络通信、硬盘及固态存储中的具体应用实例。进一步地,文章讲述了MTK-ATA技术在高
优化TI 28X系列DSP性能:高级技巧与实践(性能提升必备指南)

# 摘要
本文系统地探讨了TI 28X系列DSP性能优化的理论与实践,涵盖了从基础架构性能瓶颈分析到高级编译器技术的优化策略。文章深入研究了内存管理、代码优化、并行处理以及多核优化,并展示了通过调整电源管理和优化RTOS集成来进一步提升系统级性能的技巧。最后,通过案例分析和性能测试验证了优化
【提升响应速度】:MIPI接口技术在移动设备性能优化中的关键作用

# 摘要
移动设备中的MIPI接口技术是实现高效数据传输的关键,本论文首先对MIPI接口技术进行了概述,分析了其工作原理,包括MIPI协议栈的基础、信号传输机制以及电源和时钟管理。随后探讨了MIPI接口在移动设备性能优化中的实际应用,涉及显示和摄像头性能提升、功耗管理和连接稳定性。最后,本文展望了MIPI技术的未来趋势,分析了新兴技术标准的进展、性能优化的创新途径以及当前面临的技术挑战。本论文旨在为移动
PyroSiM中文版高级特性揭秘:精通模拟工具的必备技巧(专家操作与界面布局指南)
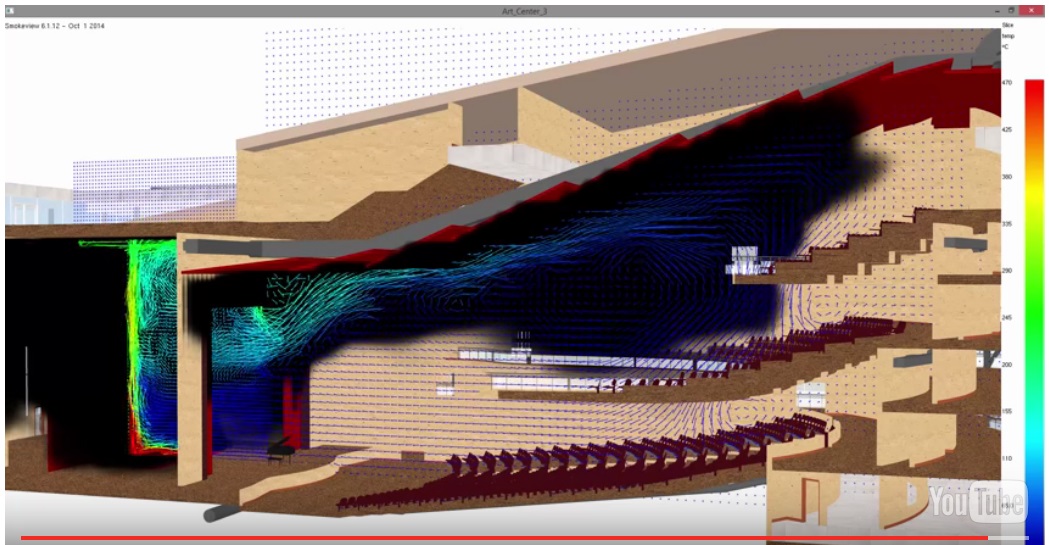
# 摘要
PyroSiM是一款功能强大的模拟软件,其中文版提供了优化的用户界面、高级模拟场景构建、脚本编程、自动化工作流以及网络协作功能。本文首先介绍了PyroSiM中文版的基础配置和概览,随后深入探讨了如何构建高级模拟场景,包括场景元素组合、模拟参数调整、环境动态交互仿真、以及功能模块的集成与开发。第三章关注用户界面的优化
【云计算优化】:选择云服务与架构设计的高效策略
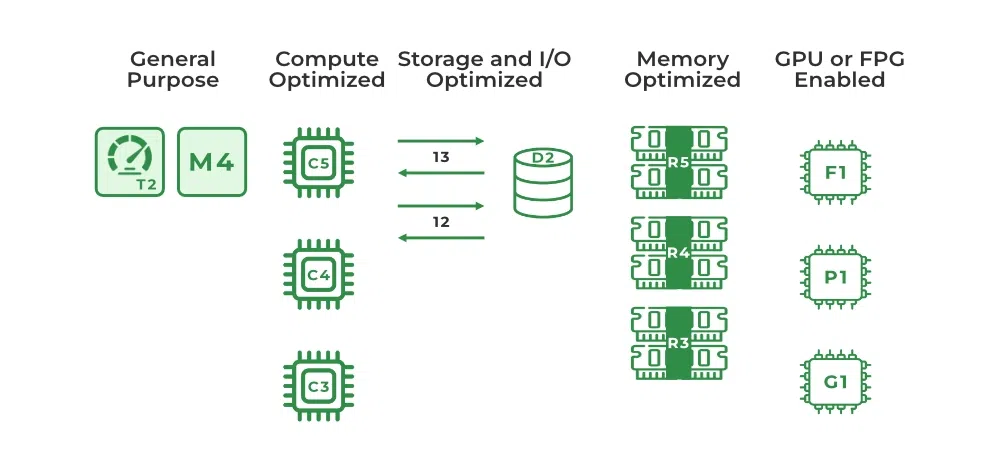
# 摘要
本文系统地探讨了云计算优化的各个方面,从云服务类型的选择到架构设计原则,再到成本控制和业务连续性规划。首先概述了云计算优化的重要性和云服务模型,如IaaS、PaaS和SaaS,以及在选择云服务时应考虑的关键因素,如性能、安全性和成本效益。接着深入探讨了构建高效云架构的设计原则,包括模块化、伸缩性、数据库优化、负载均衡策略和自动化扩展。在优化策
性能飙升指南:Adam's CAR性能优化实战案例
# 摘要
随着软件复杂性的增加,性能优化成为确保应用效率和响应速度的关键环节。本文从理论基础出发,介绍了性能优化的目的、指标及技术策略,并以Adam's CAR项目为例,详细分析了项目性能需求及优化目标。通过对性能分析与监控的深入探讨,本文提出了性能瓶颈识别和解决的有效方法,分别从代码层面和系统层面展示了具体的优化实践和改进措施。通过评估优化效果,本文强调了持续监控和分析的重要性,以实现性能的持续改进和提升。
#
【Oracle服务器端配置】:5个步骤确保PLSQL-Developer连接稳定性
# 摘要
本文对Oracle数据库服务器端配置进行了详细阐述,涵盖了网络环境、监听器优化和连接池管理等方面。首先介绍
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )