【核磁共振光谱预测】
发布时间: 2024-12-06 11:17:17 阅读量: 12 订阅数: 16 

一阶耦合图:为核磁共振光谱制作漂亮的耦合图。-matlab开发
参考资源链接:[Avogadro中文教程:分子建模与可视化全面指南](https://wenku.csdn.net/doc/6b8oycfkbf?spm=1055.2635.3001.10343)
# 1. 核磁共振光谱预测基础
## 1.1 简介与重要性
核磁共振光谱(NMR)技术是现代化学、生物化学和材料科学中不可或缺的一种分析工具。通过分析材料中原子核的磁共振信号,NMR能够提供分子结构的详细信息。NMR预测允许研究人员在没有实际实验的情况下,预测化合物的核磁共振光谱。这不仅加快了研究进程,还减少了成本,并为理解复杂体系提供了新的视角。核磁共振光谱预测是一个复杂的过程,涉及物理学、化学和计算机科学的多个方面。
## 1.2 基本概念和术语
在深入讨论NMR预测之前,了解一些基本概念和术语是必要的。谱图上的每一个峰都对应于特定类型的核在特定的化学环境中。术语如化学位移、耦合常数和积分值都是描述NMR谱图的关键参数。化学位移是指由于周围电子密度的不同,导致核感受到的磁场强度不同而产生的共振频率的微小变化。耦合常数描述了不同核之间通过化学键或空间距离产生的相互作用强度。积分值则代表了特定类型核的数量。
## 1.3 NMR预测的应用领域
NMR预测的用途广泛,从化学合成的监控到药物开发中的分子结构鉴定,再到材料科学中的高分子表征。在药物开发中,NMR预测可用于理解药物分子与靶标蛋白的相互作用。在材料科学中,NMR预测有助于研究聚合物的微观结构以及在不同条件下的动态变化。NMR预测工具的出现极大地推动了这些领域的研究进展。
# 2. 核磁共振光谱理论框架
### 2.1 核磁共振(NMR)的基本原理
#### 2.1.1 磁矩与自旋
核磁共振光谱分析是一种利用原子核在强磁场中与射频能量相互作用的特性来研究物质结构的方法。在NMR中,首先需要理解的是原子核的磁矩和自旋。原子核由质子和中子组成,具有固有的自旋量子数I。由于质子和中子自身带有磁性,因此它们赋予原子核磁矩(μ)。磁矩与自旋量子数之间的关系如下:
```
μ = γ * I * ħ
```
其中,γ是旋磁比,表示原子核对磁场的响应程度;ħ是约化普朗克常数。
磁矩的存在使得原子核在外部磁场中能够取不同的能量状态,从而为NMR信号的产生提供了基本条件。
#### 2.1.2 能级分裂与共振吸收
在外部磁场中,根据量子力学的原理,具有磁矩的原子核只能占据特定的能量级,这种现象称为能级分裂。无外部磁场时,原子核的状态是简并的,即它们具有相同的能量;而施加外部磁场后,这种简并被解除,能量级发生分裂。分裂的能量差值与外部磁场的强度成正比,与原子核的旋磁比成正比。
当施加特定频率的射频(RF)脉冲时,如果该频率与能级分裂的频率相匹配(即共振条件),原子核能够从低能级跃迁到高能级,这种跃迁对应于射频能量的吸收。由于不同类型的原子核具有不同的旋磁比,它们各自吸收特定频率的射频能量,使得可以区分不同的核类型。
### 2.2 核磁共振信号的产生
#### 2.2.1 磁场中的核自旋排列
在没有外部磁场的情况下,原子核的自旋状态是随机分布的。但是,在外部磁场中,自旋系统会趋向于达到热平衡状态,此时低能量状态的原子核数多于高能量状态的原子核数。这种不均匀的自旋排列是NMR信号产生的基础。处于高能量状态的原子核在合适的射频能量作用下,会跃迁到低能量状态,并释放能量。
#### 2.2.2 射频脉冲的应用与信号检测
射频脉冲用于激发核自旋系统,产生NMR信号。这个过程包括几个步骤:首先是射频脉冲的选择和施加,它需要准确地匹配原子核能级分裂的能量。脉冲的持续时间和强度决定了激发的效率和性质。
射频脉冲施加后,需要使用NMR探头检测弛豫过程中释放出的射频信号。弛豫是指原子核由激发态返回到平衡状态的过程,包括横向弛豫(T2)和纵向弛豫(T1)。弛豫时间对NMR信号的强度和形状有显著影响,是NMR谱图分析中的重要参数。
### 2.3 核磁共振光谱数据解析
#### 2.3.1 谱图的构成与分析方法
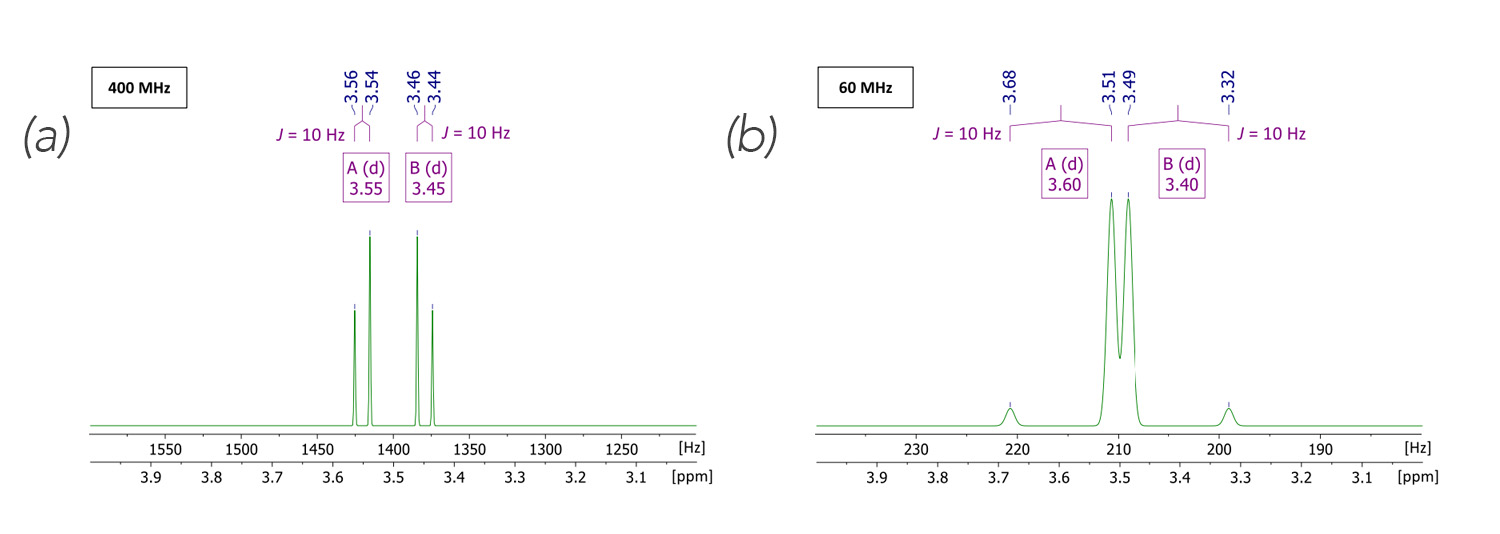
NMR谱图由一系列的共振峰组成,每个共振峰对应于一种特定的核类型在不同化学环境中所吸收的能量。通过分析共振峰的位置(化学位移)、强度、多重性(耦合常数)和面积(积分值),可以获得分子结构和动力学等信息。
化学位移反映的是原子核周围电子云的屏蔽效应,它受分子内部电子云分布的影响,因此可以用来推断分子结构。而耦合常数提供了原子核之间的相互作用信息,这种作用常发生在相邻或特定距离的原子核之间。积分值则直接关联到特定共振峰对应核的相对数量。
#### 2.3.2 化学位移、耦合常数和积分值
化学位移、耦合常数和积分值是NMR数据分析中的三个关键要素。化学位移以ppm(百万分率)为单位,它与磁场强度和特定化合物的标准值相关联。通过比较化学位移,可以确定核在分子中的化学环境。
耦合常数(J)通常以赫兹(Hz)为单位,描述的是原子核之间通过化学键传递的相互作用强度。这种相互作用可以提供关于分子几何构型的信息,尤其是在判断相邻原子核之间的耦合模式时非常有用。
积分值给出了不同共振峰下面积的相对值,反映了不同化学环境中原子核的数量比例。通过积分曲线,可以计算出具有特定化学环境的原子核数目,这对于确定分子结构中的官能团数量尤其重要。
### 2.4 核磁共振光谱数据解析进阶
#### 2.4.1 多重性分析
NMR谱图中的共振峰多重性是指共振峰按照特定模式分裂的现象。这种分裂是由于相邻的核自旋相互耦合导致的,具体表现为双峰、三峰、四峰等模式,称为双重峰、三重峰、四重峰等。多重性的分析可以提供有关核之间耦合关系的信息,进而推断分子中核的相对位置。
多重性的数目与耦合核的数量有关,遵循n+1规则,其中n是耦合的相邻核的数量。例如,一个核与两个相同类型的相邻核耦合时,会出现三重峰;与三个相同类型的相邻核耦合时,会出现四重峰。在实际应用中,多重性的分析需要结合化学位移和耦合常数来综合判断。
#### 2.4.2 二维核磁共振光谱
为了获得更为复杂分子结构的详细信息,核磁共振技术发展出了二维(2D)NMR方法。在二维NMR光谱中,通常以一个核的化学位移为横轴,另一个核的化学位移为纵轴,形成一个二维矩阵。通过分析这个矩阵,可以得到两个核之间耦合关系的空间信息,这对于确定分子的三维结构非常有帮助。
二维NMR包括多种技术,如COSY(化学移位相关光谱)、NOESY(核Overhauser效应光谱)、HSQC(异核单量子相干光谱)等。每种技术有其特定的应用范围和优势,例如COSY适用于分析相邻核之间的耦合关系,而NOESY可以提供核之间通过空间距离耦合的信息。
### 2.5 小结
本章从基础理论角度介绍了核磁共振光谱的基本原理和信号产生机制,进一步解析了NMR数据的基本组成元素:化学位移、耦合常数和积分值,并引入了二维核磁共振光谱的高级技术。通过这些内容的介绍,我们为读者搭建起了一个理解NMR光谱预测的理论框架,为下一章中探讨预测模型的构建奠定了基础。
# 3. 预测模型的构建与优化
## 3.1 量子化学计算在NMR预测中的应用
### 3.1.1 从头算和密度泛函理论
从头算(Ab initio)方法和密度泛函理论(DFT)是量子化学计算的两种主要方法,它们在核磁共振(NMR)预测中扮演着重要的角色。从头算方法基于量子力学原理,通过精确求解薛定谔方程来获取分子的电子结构信息。然而,随着分子体系的增大,所需的计算资源和时间会呈指数级增长,限制了从头算在较大分子体系中的应用。
相比之下,密度泛函理论(DFT)基于电子密度而不是波函数来描述电子系统,这大大减少了所需的计算资源和时间。DFT方法在NMR预测中的广泛应用得益于其在计算效率和准确性之间的良好平衡。它能够以相对较少的计算代价,提供准确的化学位移预测,尤其适合于中等大小的有机分子。
在实际应用中,科学家通常会根据研究对象的特性以及计算资源的可用性,选择合适的计算方法。从头算方法可能更多地用于小分子的精确计算,而DFT则适用于更复杂的生物大分子和固体材料的NMR预测。
### 3.1.2 计算方法的选择与参数设定
选择合适的计算方法和设定准确的计算参数是实现有效NMR预测的关键步骤。在量子化学计算中,关键的参数包括基组选择、泛函类型、溶剂效应模型等。
基组是指用于描述分子轨道的一组基函数。不同类型的基组在计算精度和效率上有所不同,如STO-3G、6-31G、cc-pVDZ等。基组的选择需考虑到分子的大小、目标计算精度以及可用的计算资源。
泛函类型的选择也对计算结果影响巨大。不同的泛函在处理电子相关问题上的表现各异,常见的泛函如B3LYP、PBE、M06等。科研人员需根据具体问题选择合适的泛函,以达到最优化的预测效果。
最后,溶剂效应是影响NMR化学位移的重要因素之一,特别是在溶液NMR实验中。因此,合理的溶剂效应模型,如极化连续介质模型(PCM)或自洽反应场(SCRF),也是实现准确预测的关键。
代码块示例:
```bash
# 示例计算脚本,使用Gaussian09软件进行DFT计算
%Chk=myfile
%Mem=4GB
#P B3LYP/6-31G(d) SCRF=(Solvent=
```

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
PyroSiM中文版模拟效率革命:8个实用技巧助你提升精确度与效率
# 摘要
PyroSiM是一款强大的模拟软件,广泛应用于多个领域以解决复杂问题。本文从PyroSiM中文版的基础入门讲起,逐渐深入至模拟理论、技巧、实践应用以及高级技巧与进阶应用。通过对模拟理论与效率提升、模拟模型精确度分析以及实践案例的探讨,本文旨在为用户提供一套完整的PyroSiM使用指南。文章还关注了提高模拟效率的实践操作,包括优化技巧和模拟工作流的集成。高级
QT框架下的网络编程:从基础到高级,技术提升必读
# 摘要
QT框架下的网络编程技术为开发者提供了强大的网络通信能力,使得在网络应用开发过程中,可以灵活地实现各种网络协议和数据交换功能。本文介绍了QT网络编程的基础知识,包括QTcpSocket和QUdpSocket类的基本使用,以及QNetworkAccessManager在不同场景下的网络访问管理。进一步地,本文探讨了QT网络编程中的信号与槽
优化信号处理流程:【高效傅里叶变换实现】的算法与代码实践

# 摘要
傅里叶变换是现代信号处理中的基础理论,其高效的实现——快速傅里叶变换(FFT)算法,极大地推动了数字信号处理技术的发展。本文首先介绍了傅里叶变换的基础理论和离散傅里叶变换(DFT)的基本概念及其计算复杂度。随后,详细阐述了FFT算法的发展历程,特别是Coo
MTK-ATA核心算法深度揭秘:全面解析ATA协议运作机制

# 摘要
本文深入探讨了MTK-ATA核心算法的理论基础、实践应用、高级特性以及问题诊断与解决方法。首先,本文介绍了ATA协议和MTK芯片架构之间的关系,并解析了ATA协议的核心概念,包括其命令集和数据传输机制。其次,文章阐述了MTK-ATA算法的工作原理、实现框架、调试与优化以及扩展与改进措施。此外,本文还分析了MTK-ATA算法在多
【MIPI摄像头与显示优化】:掌握CSI与DSI技术应用的关键
# 摘要
本文全面介绍了MIPI摄像头与显示技术,从基本概念到实际应用进行了详细阐述。首先,文章概览了MIPI摄像头与显示技术的基础知识,并对比分析了CSI与DSI标准的架构、技术要求及适用场景。接着,文章探讨了MIPI摄像头接口的配置、控制、图像处理与压缩技术,并提供了高级应用案例。对于MIPI显示接口部分,文章聚焦于配置、性能调优、视频输出与图形加速技术以及应用案例。第五章对性能测试工具与
揭秘PCtoLCD2002:如何利用其独特算法优化LCD显示性能

# 摘要
PCtoLCD2002作为一种高性能显示优化工具,在现代显示技术中占据重要地位。本文首先概述了PCtoLCD2002的基本概念及其显示性能的重要性,随后深入解析了其核心算法,包括理论基础、数据处理机制及性能分析。通过对算法的全面解析,探讨了算法如何在不同的显示设备上实现性能优化,并通过实验与案例研究展示了算法优化的实际效果。文章最后探讨了PCtoLCD2002算法的进阶应用和面临
DSP系统设计实战:TI 28X系列在嵌入式系统中的应用(系统优化全攻略)
# 摘要
TI 28X系列DSP系统作为一种高性能数字信号处理平台,广泛应用于音频、图像和通信等领域。本文旨在提供TI 28X系列DSP的系统概述、核心架构和性能分析,探讨软件开发基础、优化技术和实战应用案例。通过深入解析DSP系统的设计特点、性能指标、软件开发环境以及优化策略,本文旨在指导工程师有效地利用DSP系统的
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )