开源软件助力氢键计算模拟:应用与挑战的全面解读
发布时间: 2024-12-13 17:40:12 阅读量: 2 订阅数: 8 

jOcular:用于模拟镜头,棱镜等系统的光学设计软件。-开源

参考资源链接:[Materials Studio教程:计算与显示氢键](https://wenku.csdn.net/doc/733iggjso3?spm=1055.2635.3001.10343)
# 1. 开源软件在计算模拟中的角色
随着科技的不断进步,计算模拟已经成为科学研究中不可或缺的工具。尤其在物理、化学、生物等领域,计算模拟能够提供实验难以达到的微观视角。在这一过程中,开源软件扮演着至关重要的角色。
开源软件的优势在于其透明性和可扩展性,这使得全球的研究者们能够在前人的基础上进行创新,共同推动技术的发展。此外,开源软件通常伴随着活跃的社区支持,研究者们可以通过社区交流、分享代码和经验,快速解决遇到的问题。
本章将深入探讨开源软件在计算模拟中的应用,包括它如何影响科研进程,以及如何选择适合特定研究需求的软件工具。我们还将讨论开源软件如何促进科研的民主化和开放性,以及它在实现科学突破方面所起到的积极作用。通过开源软件,研究者们可以节省成本,提高效率,并且享受到来自全球社区的帮助与合作。
# 2. ```
# 第二章:氢键的基本理论和计算方法
在现代化学和生物科学中,氢键作为分子间作用力的一种,发挥着至关重要的作用。氢键的存在不仅影响分子的几何结构和构象稳定性,还在DNA复制、蛋白质折叠等生物过程中扮演着不可或缺的角色。本章将深入探讨氢键的基础知识、计算模拟方法以及如何选择合适的软件进行氢键的模拟研究。
## 2.1 氢键的基础知识
### 2.1.1 氢键的定义和特性
氢键是比共价键弱、比范德华力强的一种偶极相互作用。它通常发生在含有电负性强的原子(如氧、氮)与氢原子之间,当一个原子的氢原子与另一个电负性强的原子接近时,便形成了氢键。氢键的键能一般在4到40千焦/摩尔之间。
在分子层面,氢键不仅能够影响分子的稳定性,还能够控制分子间的识别和结合,例如,在水分子间就存在大量的氢键作用,使得水具有异常的热容和沸点。
### 2.1.2 氢键在分子结构中的作用
氢键对于大分子结构的稳定性至关重要,它有助于蛋白质二级结构的形成,比如α螺旋和β折叠。在DNA分子中,氢键在碱基对之间起着粘合剂的作用,对遗传信息的稳定传递至关重要。
## 2.2 氢键计算的方法论
### 2.2.1 经典力场方法
经典力场方法是模拟氢键和其它分子间作用力的常用方法。它基于经典的牛顿力学原理,利用经验或半经验公式来描述原子间相互作用势能,以预测分子体系的行为。
在氢键的模拟中,力场方法的优势在于速度快、计算成本低,因此非常适用于大规模模拟。但是,这种方法的局限性在于它依赖于参数化的准确性和适用性,对于复杂的化学环境可能缺乏足够的精确性。
### 2.2.2 量子化学计算方法
量子化学计算方法则是基于量子力学原理来计算分子的电子结构和能量。相对于经典力场方法,量子化学可以提供更精确的氢键描述,特别是在涉及电子重新分布的化学反应中。
氢键的量子化学计算通常涉及波函数理论或密度泛函理论,能够在电子层面上精确模拟氢键的形成和断裂过程。然而,量子化学计算的代价是高昂的计算资源需求和复杂的计算过程。
## 2.3 氢键模拟软件的选择标准
### 2.3.1 软件的计算精度和效率
在选择氢键模拟软件时,首先要考虑的是其计算精度和效率。高精度的软件可以提供更加可靠的模拟结果,但在复杂系统中,这往往意味着需要较长的计算时间。
一些软件例如Gaussian、NWChem和Q-Chem等,在处理特定类型的量子化学计算时,提供了很好的精度和效率的平衡。
### 2.3.2 软件的开源社区和用户支持
对于氢键模拟这类需要高度定制和优化的计算过程,强大的开源社区和良好的用户支持是选择软件时不可忽视的因素。开源软件如CP2K、SIESTA等,不仅提供源代码,还有活跃的社区来帮助解决遇到的问题。
## 代码块示例及注释
在氢键的模拟计算中,使用量子化学计算软件Gaussian进行一个简单的分子结构优化示例,Gaussian使用输入文件(通常以`.gjf`为扩展名)来进行计算。
```gjf
%NProcShared=4
%Mem=4GB
#opt freq b3lyp/6-31G(d,p) geom=connectivity
Molecule with Hydrogen Bonding
0 1
C 0.000000 -0.000000 -0.127891
O 0.000000 0.000000 1.114258
H 0.000000 -0.820594 -0.583711
H 0.000000 0.820594 -0.583711
N 1.082459 0.000000 -1.080465
H 1.926965 -0.000000 -0.560262
H 1.070550 -0.000000 -2.066187
```
以上是使用Gaussian软件进行氢键模拟的一个基础输入文件示例。代码段中的`%NProcShared=4`和`%Mem=4GB`设置分别指定了使用4个处理器核心和4GB的内存。`#opt freq b3lyp/6-31G(d,p) geom=connectivity`行定义了具体的计算任务,即使用B3LYP密度泛函和6-31G(d,p)基组进行几何优化和频率计算。
在执行上述输入文件之前,用户需要确保Gaussian软件已经被正确安装在计算平台上,然后在命令行中运行该输入文件。例如,在Linux系统中,执行以下命令:
```bash
g16 < input.gjf > output.log
```
这样,Gaussian软件会根据输入文件中的设置进行计算,并将结果输出到`output.log`文件中。计算完成后,用户可以分析输出文件来获取关于氢键的能量、几何构型以及振动频率等信息。
## 表格示例
在进行氢键模拟研究时,研究人员通常需要对比不同方法的优势和局限性。以下是一个表格示例,展示了几种常见的氢键模拟方法及其特点:
| 模拟方法 | 优点 | 局限性 | 典型软件 |
|-----------|------|---------|------------|
| 经典力场 | 计算速度快,适用于大规模模拟 | 参数化依赖性强,可能缺乏精确性 | AMBER, GROMACS |
| 密度泛函理论 | 精确模拟电子层面相互作用 | 计算资源需求高,处理复杂系统时效率低 | Gaussian, Q-Chem |
| 从头算方法 | 理论上精确,适用于小分子模拟 | 高成本和复杂性,不适用于大型体系 | NWChem, CFOUR |
在上述表格中,我们可以清楚地看到不同模拟方法在氢键研究中的应用和局限。例如,经典力场方法在处理大型生物分子时具有速度快和资源消耗低的优势,但在参数化准确性方面存在缺陷。而量子化学方法虽然提供了精确的结果,却在处理复杂系统时遇到了计算资源和效率的挑战。
通过对比不同模拟方法的优缺点,研究人员可以根据自己的研究目标和条件,选择最合适的计算模拟工具来进行氢键研究。这种基于实际需求的方法论选择,对于提升研究效率和准确性具有重要意义。
## mermaid 流程图示例
为了更直观地展示氢键模拟的计算流程,以下是一个使用mermaid语法的流程图示例。该流程图详细描述了使用量子化学软件进行氢键计算分析的步骤。
```mermaid
flowchart LR
A[开始] --> B[输入文件准备]
B --> C{输入文件检查}
C -->|正确| D[提交计算任务]
C -->|错误| E[修改输入文件]
E --> B
D --> F[监控计算过程]
F -->|完成| G[结果分析]
F -->|错误或中断| H[

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏重点介绍材料工作室中氢键显示方法的创新应用,以及人工智能在氢键设计中的前沿探索。通过人工智能技术,研究人员能够更深入地了解氢键的性质,并预测新材料的性能。专栏文章涵盖了氢键在药物设计、催化、能源储存和电子设备等领域中的重要性。通过结合材料工作室的强大模拟能力和人工智能的预测能力,研究人员可以设计出具有增强功能和改进性能的新材料。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
台电平板双系统维护宝典:备份、更新与性能优化技巧
# 摘要
本文介绍了台电平板双系统的操作与维护,首先概述了双系统的基本概念,随后详述了备份策略与技巧,重点在于不同备份方法的实施与实践操作。进一步,文章探讨了双系统更新与故障修复的机制、监控与性能优化方法。此外,本文还探讨了系统维护中的高级技巧,如系统定制、性能优化和安全性加固。最后,通过案例分析综合应用章节,对双系统的维护工具与资源进行了推荐,并对维护的未来趋势进行了展望。整体而言,本文为台电平板用户提供了全面的双系统管理知识和高级技巧,旨在提高用户对平板双系统的操作效率与安全性。
# 关键字
台电平板;双系统;数据备份;系统更新;故障诊断;性能优化;系统维护
参考资源链接:[台电平板双
【水利项目效率提升】:HydrolabBasic应用案例深度剖析

# 摘要
HydrolabBasic是一款集成了先进水文数据分析、流量估算、洪水预报及水质监测功能的软件,旨在优化水资源管理和提高水利项目的决策支持。本文介绍了HydrolabBasic的基础理论、核心算法及其在实际水利项目中的应用,如水资源规划、洪水监测预警和水质保护。文章还探讨了软件的高级功能,
揭秘CAN总线架构:从原理到工业应用的全面解析
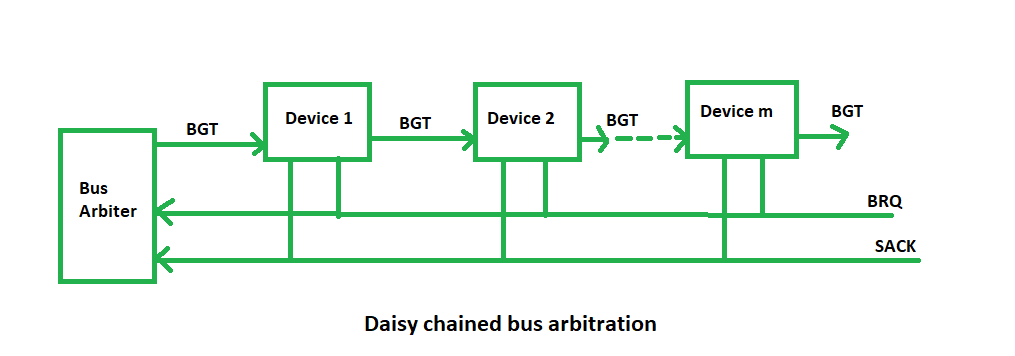
# 摘要
本文系统地介绍了CAN总线的基础理论、协议细节、硬件实现以及在工业自动化中的应用。文章首先阐述了CAN总线的起源、发展及协议标准,分析了数据帧结构、传输机制和网络中的消息仲裁过程。随后,深入讨论了CAN控制器和收发器的工作原理,以及网络布线、电气特性和故障诊断方法。文章还探讨了CAN总线在工业自动化中的实际应用,包括与工业现场总线标准的集成、实时性能的需求,以及安全性与可靠性方面的考虑。最后,展望了CAN总线
【XJC-608T-C控制器高级设置】:优化Modbus通讯性能(性能提升全攻略)
# 摘要
本文详细介绍了XJC-608T-C控制器的Modbus通讯性能优化过程。首先,对控制器和Modbus通讯协议进行了概述,阐述了Modbus协议架构及性能理论基础。接着,探讨了影响Modbus通讯性能的关键因素,包括网络延迟、设备处理能力及信号干扰,并提供了理论上的性能优化方法。文中进一步阐释了XJC-608T-C控制器的高级设置步骤和原则,以及通讯参数的调优策略。通过实践案例分析,本文展示了在不同工业应用场景下对通讯性能进行提升的具体操作步骤、测试与监控,以及之后的维护和优化。最后,总结了性能优化经验,并对通讯技术的未来趋势进行了展望,特别是针对XJC-608T-C控制器的应用前景。
STM32F4内存管理优化:程序与数据存储的高级策略

# 摘要
本文深入探讨了STM32F4微控制器的内存管理机制及其优化策略。首先,概述了STM32F4的基础内存概念和结构,强调了内存管理单元(MMU)与内存保护单元(MPU)的作用。接着,分析了程序存储优化的关键策略,包括静态与动态内存分配、堆栈管理以及编译器优化选项。在数据存储方面,本文探讨了常量、全局变量的内存布局、数据缓存和缓冲机制,以及DMA数据传输的优化。通过实践案例分析,文章提
Layui Table列自定义内容显示:图片展示的最佳实践
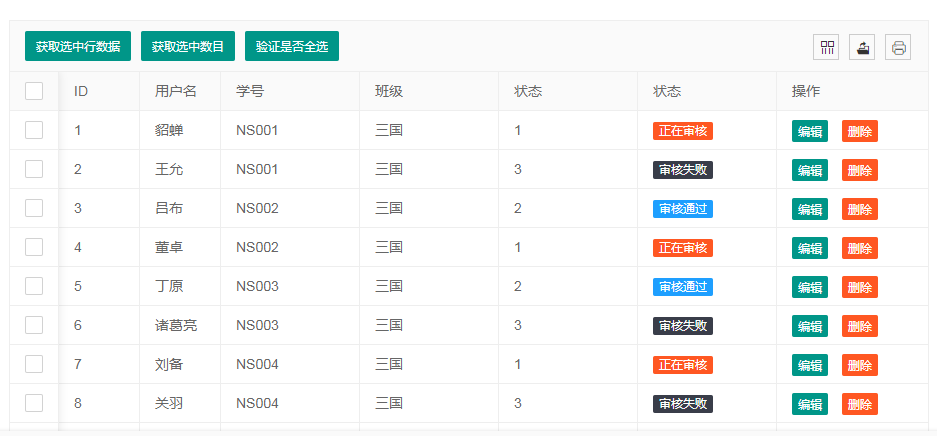
# 摘要
本文详细介绍了Layui Table组件的基础知识及其列自定义显示技术。首先概述了Layui Table的基本概念和必要的列配置方法,随后深入探讨了前端显示技术在列自定义内容显示中的应用,包括HTML/CSS/JavaScript以及图片展示技术的原理与实现。接着,文章通过实践案例阐述了如何实现基础与高级的图片展示功能,并关注了交互优化的实施。进阶应用部分着重讲述
从零开始掌握MapReduce:学生成绩统计编程模型详解
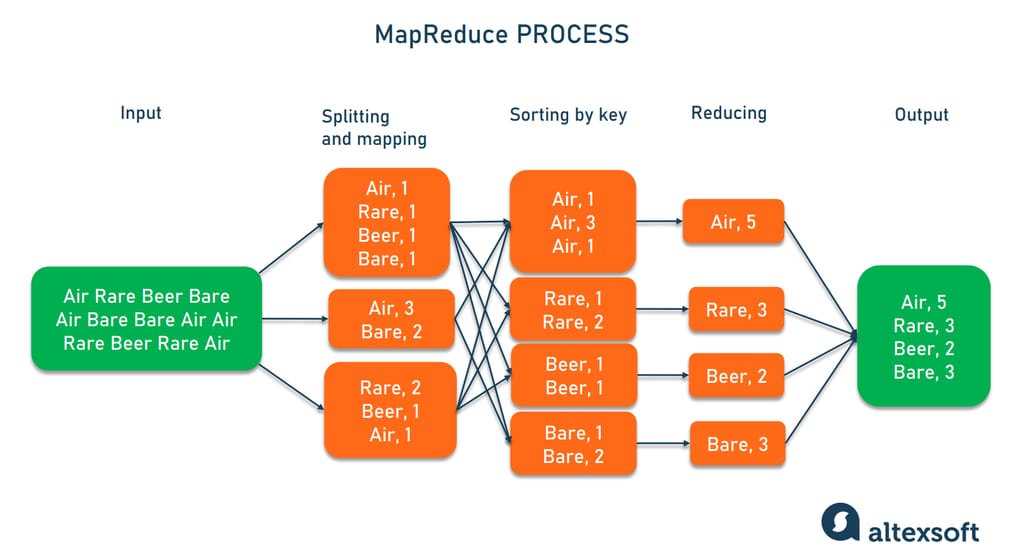
# 摘要
MapReduce作为一种编程模型,广泛应用于大规模数据处理。本文首先概述了MapReduce编程模型的基本概念,然后深入探讨了其核心理论与机制,包括计算模型、数据流、任务调度和容错机制。接着,文章通过实战入门篇指导读者搭建编程环境、编写基本的MapReduce程序,以及实现具体案例。此外,本文详细分析了MapReduce在学生成绩统计
三菱FX3U PLC终极指南:硬件连接、USB通信与故障排除(全方位解读手册)

# 摘要
本文详细介绍了三菱FX3U PLC的基础知识、硬件连接、USB通信设置、程序开发与调试、故障诊断与排除,以及在工业自动化应用中的案例和新技术展望。通过对PLC硬件组件的解析、电源接线指导以及端口配置的讲解,文章为读者提供了全面的硬件配置知识。USB通信章节则探讨了通信基础、配置步骤和实际操作中
光盘挂载控制环路设计最佳实践:实现高效稳定的黄金法则
# 摘要
本文主要探讨了光盘挂载控制环路的设计与实现,从理论基础到实践应用,再到未来的发展展望进行了全面的分析和讨论。首先介绍了光盘挂载控制的基本概念、目标和原则,进而阐述了关键参数的定义及其对系统性能的影响,以及系统稳定性理论的分析。随后,文章深入到实践层面,详细讲解了挂载控制环路的设计、测试、优化以及故障处理和维护策略。
MT6825编码器:如何通过精确校准确保最佳性能?
# 摘要
MT6825编码器是精密测量和控制领域的重要设备,本文首先介绍了其基本工作原理和性能指标,随后深入探讨了精确校准的理论基础,包括性能指标解析、校准方法、技术和工具。文章第三章详细叙述了MT6825编码器的校准实践流程,从准备到执行校准,再到校准后的验证与调整步骤。接着,本文对编码器进行了优化与故障排除分析,提供了实用的案例和故障排除技巧。此外,本文还探讨了MT6825编码器在工业自动化、测试与测量以及特殊环境下的多样化应用。最后一章展望了编码器技术的发展趋势,分析了新技术和行业需求对编码器性能和应用的潜在影响,以及面对未来挑战的战略规划。
# 关键字
MT6825编码器;校准理论;
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )