量子化学前沿技术:氢键计算解析
发布时间: 2024-12-13 16:44:24 阅读量: 2 订阅数: 8 

基于net的超市管理系统源代码(完整前后端+sqlserver+说明文档+LW).zip
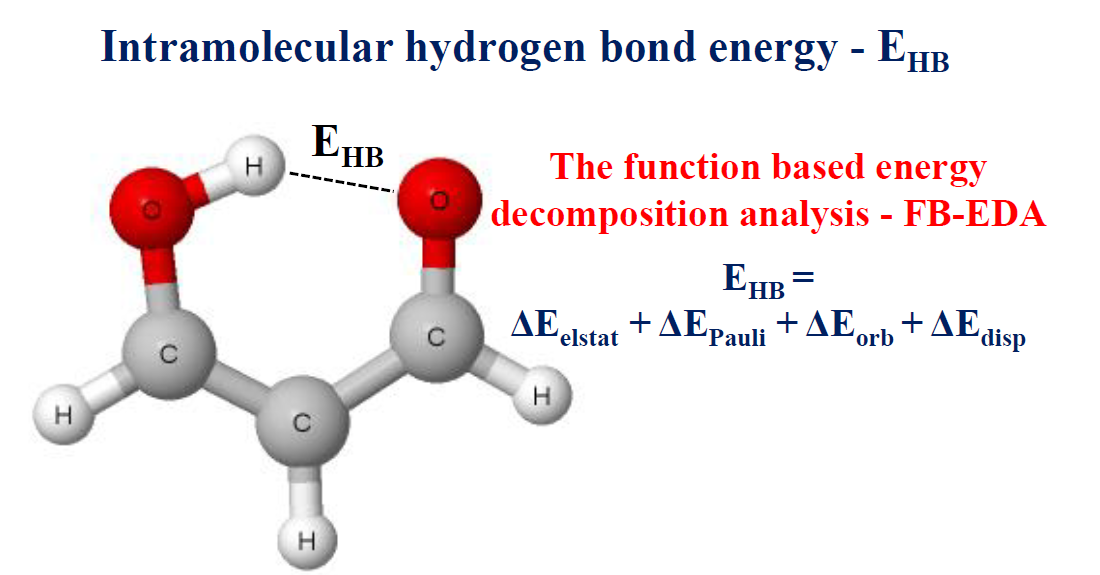
参考资源链接:[Materials Studio教程:计算与显示氢键](https://wenku.csdn.net/doc/733iggjso3?spm=1055.2635.3001.10343)
# 1. 量子化学与氢键基础
## 1.1 氢键的定义和重要性
氢键是自然界中最常见的一种弱相互作用,它在分子生物学、材料科学及药物设计等领域中发挥着不可替代的作用。氢键能够影响分子的稳定性和空间结构,从而决定物质的物理化学性质。本章将深入浅出地讲解氢键的基本概念,为后续章节关于氢键计算理论和软件工具的探讨打下坚实基础。
## 1.2 量子化学简述
量子化学是应用量子力学原理研究分子和化学体系的科学。它为我们理解原子、分子以及它们之间的相互作用提供了数学模型和理论框架。在氢键研究中,量子化学的应用尤为重要,因为它能够提供分子内部电子分布和相互作用的精确描述。
## 1.3 氢键与量子化学的交点
量子化学和氢键研究的交汇点在于对氢键量子特性的深入理解。例如,氢键的形成、强度和方向性都可以通过量子化学方法来模拟和解释。这些理解和分析对于预测和设计氢键驱动的新材料和药物分子至关重要。本章将通过量子力学的视角,为读者打开理解氢键的新窗口。
# 2. 氢键计算的理论基础
## 2.1 量子力学在氢键研究中的应用
### 2.1.1 量子化学的基本原理
量子化学是化学的一个分支,它使用量子力学的原理来解释和预测分子的性质和反应。在氢键研究中,量子化学能够提供分子内部电子分布的详细图像,这对于理解氢键的形成机制至关重要。在量子化学中,氢键被视为电子云重叠和极性相互作用的结果。
量子化学的基本原理涉及波函数的构建和薛定谔方程的求解。波函数描述了粒子的量子态,而薛定谔方程则是用来找到这个波函数的数学表达式。对于多电子系统,如水分子,精确求解薛定谔方程是非常复杂的。因此,通常采用近似方法,如Hartree-Fock方法、密度泛函理论等。
波函数和电子密度在氢键分析中扮演关键角色。例如,通过波函数可以确定电子在分子内的分布情况,进一步分析两个分子间的电荷转移以及形成的偶极矩,这些都是氢键形成和稳定性的关键因素。
### 2.1.2 氢键的量子描述
氢键是一种弱的化学键,它在形成时通常涉及一个带部分正电荷的氢原子和一个带负电的原子(如氧、氮或氟)。在量子力学框架下,氢键可以被视为一种通过偶极相互作用产生的吸引力。
为了精确描述氢键,研究人员通常依赖于量子化学的计算方法。这些方法包括使用波函数对体系能量的评估,波函数的优化以及多体相互作用的分析。在计算氢键时,一个重要的步骤是确定体系的基态,这通常通过求解优化后的Hartree-Fock方程或使用密度泛函理论(DFT)来实现。
氢键的量子描述不仅仅局限于静态的键强度分析,也涉及到反应动力学和热力学性质的探究。量子化学提供了一套工具来模拟氢键在不同环境(如溶剂环境、温度变化)下的行为,这对于理解生物分子和材料中的氢键作用尤为关键。
## 2.2 分子模拟技术
### 2.2.1 分子动力学模拟
分子动力学模拟是通过计算分子运动和相互作用来研究物质性质的一种技术。在这个模拟中,分子被视为由原子构成的粒子,通过牛顿运动定律来模拟它们的行为。分子动力学模拟允许研究者在原子级别上探究分子的动态性质,如扩散系数、粘度、热导率等。
在氢键研究中,分子动力学模拟可以用来模拟溶剂环境下的氢键动力学过程,比如水分子之间的氢键网络是如何在不同温度和压力下变化的。一个典型的分子动力学模拟流程包括初始化系统配置、设定力场、进行能量最小化、平衡和生产运行等步骤。
```mermaid
graph TD
A[开始模拟] --> B[初始化系统配置]
B --> C[设定力场]
C --> D[能量最小化]
D --> E[平衡阶段]
E --> F[生产运行]
F --> G[数据分析]
G --> H[结束模拟]
```
模拟过程中,力场参数的选择对结果准确性有决定性影响。力场是一套参数和方程,用来描述原子间的相互作用势能。常见的力场有AMBER、CHARMM和OPLS等。
### 2.2.2 第一性原理计算
第一性原理计算也称为从头计算,它基于量子力学的基本原理,无需依赖经验参数来计算材料的电子性质和结构。这种方法特别适合于氢键的研究,因为它可以提供精确的电荷分布和键级分析。
在第一性原理计算中,最常用的量子化学方法是密度泛函理论(DFT)。DFT将多电子波函数问题转化为电子密度问题,从而大大简化了计算过程,但仍然能够保持较高的准确性。DFT计算中最重要的部分是选择合适的交换-相关泛函。交换-相关泛函决定了电子之间的交换和相关作用,影响计算结果的准确性。
第一性原理计算适用于研究氢键在固体材料、催化剂、分子吸附等过程中的行为。它能够提供深入的物理洞察,帮助理解氢键在分子间作用力中的角色。
### 2.2.3 蒙特卡罗方法在氢键分析中的应用
蒙特卡罗方法是一种统计模拟技术,广泛应用于物理、化学以及工程学等领域。它通过随机抽样和概率统计方法来模拟系统的行为,特别适用于处理复杂的多体系统,如生物大分子和高分子材料。
在氢键分析中,蒙特卡罗方法可以用来估算体系的热力学性质,比如自由能、熵和焓等。通过随机移动分子和原子,可以模拟分子的构象变化,从而分析氢键对体系稳定性的影响。由于蒙特卡罗模拟不需要时间步长的概念,它可以用于长周期的静态或动态平均性质计算。
蒙特卡罗模拟的一个主要优点是能够处理具有多个局部最小能态的复杂体系。在氢键研究中,这种能力使得蒙特卡罗方法能够揭示氢键网络的多样性和灵活性。
## 2.3 氢键的能量和几何特性
### 2.3.1 氢键的键能计算
氢键的键能计算是指对氢键相互作用的强度进行量化的过程。键能是描述分子间相互作用力强度的重要物理量,它对于理解分子结构和反应机制至关重要。
氢键的键能通常通过量子化学计算方法来估算,如Hartree-Fock、Møller-Plesset微扰理论(MP2)、以及各种密度泛函理论方法。计算时,键能可以通过键断裂的能量差来得到,即从体系总能量中减去未形成氢键时的能量。
在实际操作中,量子化学计算软件(如Gaussian、ORCA)可以通过定义一个或多个化学键断裂步骤,计算出氢键的键能。这个过程涉及对反应物、产物以及过渡态的几何优化和频率分析。
```markdown
### 代码示例:Gaussian 16 中的氢键键能计算
**输入文件:hydrogen_bond-energy.gjf**
```gjf
%Chk=hydrogen_bond-energy
#P B3LYP/6-311++G(2d,2p) Opt freq
Title Card for Hydrogen Bond Energy Calculation
O 0.000000 0.000000 0.000000
H 0.000000 0.000000 0.988930
F -0.896851 -0.621157 1.053877
```
**解释:**
- `%Chk=hydrogen_bond-energy` 定义了检查点文件名。
- `#P B3LYP/6-311++G(2d,2p) Opt freq` 指定了计算方法和水平,进行了几何优化(Opt)和频率分析(freq)。
- 输入文件中的坐标定义了氢键体系的初始几何构型。
通过优化计算后,可以得到氢键体系的键能值。这通常涉及到输出文件中对于总能量和部分键断裂能量的分析。
```

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏重点介绍材料工作室中氢键显示方法的创新应用,以及人工智能在氢键设计中的前沿探索。通过人工智能技术,研究人员能够更深入地了解氢键的性质,并预测新材料的性能。专栏文章涵盖了氢键在药物设计、催化、能源储存和电子设备等领域中的重要性。通过结合材料工作室的强大模拟能力和人工智能的预测能力,研究人员可以设计出具有增强功能和改进性能的新材料。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
尾差结转的秘密:10分钟掌握生产成本中心的优化策略

# 摘要
生产成本中心是企业管理中的重要组成部分,它关系到企业生产活动的成本控制和效率提升。本文首先概述了生产成本中心的概念和重要性,随后详细探讨了其理论基础,包括成本中心的定义、功能以及与利润中心的区分,还有生产成本的分类和核算方法。此外,本文还分析了成本中心的建立和优化策略,以及尾差结转的原理、应用和风险管理。最后,本文通过案例展示了生产成本中心优化策略的实施、效果评估和持续改进的实际操作,以提高生产
【性能王者】:用Navicat for Oracle打造极致高效的Oracle数据库
# 摘要
本论文系统介绍了Oracle数据库的基础知识以及Navicat工具的使用。首先详细讲解了Navicat for Oracle的安装、配置流程和用户权限管理。随后,重点阐述了高效数据库管理技巧,包括对象管理、数据操作、同步策略以及性能监控与调优。接着,针对Oracle数据库性能优化,深入探讨了索引优化、查询优化和并发控制的策略。第五章介绍了自动化管理、备份与恢复以及数据分析的高级功能。最后,通过案例研究,展示了Navicat for Oracle在实际问题解决中的应用,包括大数据量处理、数据库安全性和性能瓶颈优化。本文旨在为数据库管理员提供实践指导,提升Oracle数据库的管理效率和性
【电动车仪表快速修复】:电路故障的即时识别与解决方法

# 摘要
本文全面探讨了电动车仪表的基本概念、功能、电路故障理论基础以及故障的即时识别技术。文中详细阐述了电路故障的类型、检测原理和故障诊断流程,同时提供了电动车仪表故障的识别、诊断与修复方法,强调了仪表板显示异常、电源故障和传感器信号故障的处理。文章进一步介绍了仪表的实践修复操作,包括组件更换与修理、线路修复与重接技术以及集成电路故障的修复。最后,本文讨论了电动车仪表的
SW3518S功耗管理秘籍:寄存器调整实现最佳效能
+to+enable+clock+to+I2C.jpg)
# 摘要
本文系统地介绍了SW3518S的功耗管理策略和实践技巧。首先,概述了SW3518S功耗管理的基本概念及寄存器基础知识,重点解析了寄存器在功耗控制中的作用和配置方法。随后,深入探讨了静态和动态功耗优化方法,并提出了具体的实践技巧。进阶应用章节分析了如何在保持性
【日本兄弟钻攻中心D00:新手必备10大操作指南】

# 摘要
本文详细介绍了日本兄弟钻攻中心D00的操作与维护,包括机器的安装布局、软件操作界面熟悉、工具与夹具的正确使用、编程与自动操作技巧,以及故障的诊断与解决。通过系统地阐述设备的初步操作流程、刀具与夹具的选择及应用、程序的测试与优化方法,本文旨在为操作人员提供一个全面的操作指导。文章还分享了高级应用技术、工程案例分析,并展望了未来技术发展趋势,强调了精确加工技术和复杂曲面加工技术的重要性。最后,本文探讨了行业
【Vivado实战攻略】:构建高性能视频字符叠加系统的完整指南

# 摘要
本文详细阐述了基于Vivado设计套件的视频字符叠加系统的开发流程,涵盖了系统架构设计、视频处理、字符渲染、系统级性能优化等方面。通过对FPGA资源分配、视频处理单元设计原理、IP核集成、字符叠加算法以及VHDL/Verilog语言的应用进行深入分析,文章展示了字符叠加功能的实现与优化。随后,文章转入Vivado项目实战,详细介绍了字符叠加系统的开发流程,包括项目初始化、模
高解析音频流革命家:TX-NR545流媒体支持全面解读

# 摘要
本文详细探讨了TX-NR545流媒体设备的功能和应用,从其支持的高解析音频格式到音频处理技术,再到多房间音乐流功能,提供了全面的技术解析。文章还涉及了如何实践TX-NR545的流媒体功能,包括网络设置、连接流媒体服务以及同步和延迟问题的处理。此外,本文还介绍了高级配置和优化技巧,包括音频设置和固
Android持续运行技巧:前台服务与通知的高级用法指南
# 摘要
本文系统性地探讨了Android前台服务与通知的机制、实践和结合应用。文章首先概述了前台服务与通知的基本概念和重要性,随后深入分析前台服务的工作原理、创建和管理过程,以及通知的结构与交互。通过高级应用实例,如音频播放器和实时位置追踪,文章展示了前台服务与通知如何进行有效结合,并提出优化和性能提升的方法。案例分析部分提供了实际应用场景分析和问题排查解决策略,最后展望了Androi
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )