R语言dbscan聚类:一次掌握20大核心技巧和高级应用
发布时间: 2024-11-03 16:18:11 阅读量: 32 订阅数: 37 

dbscan1d:DBSCAN聚类算法的有效一维实现
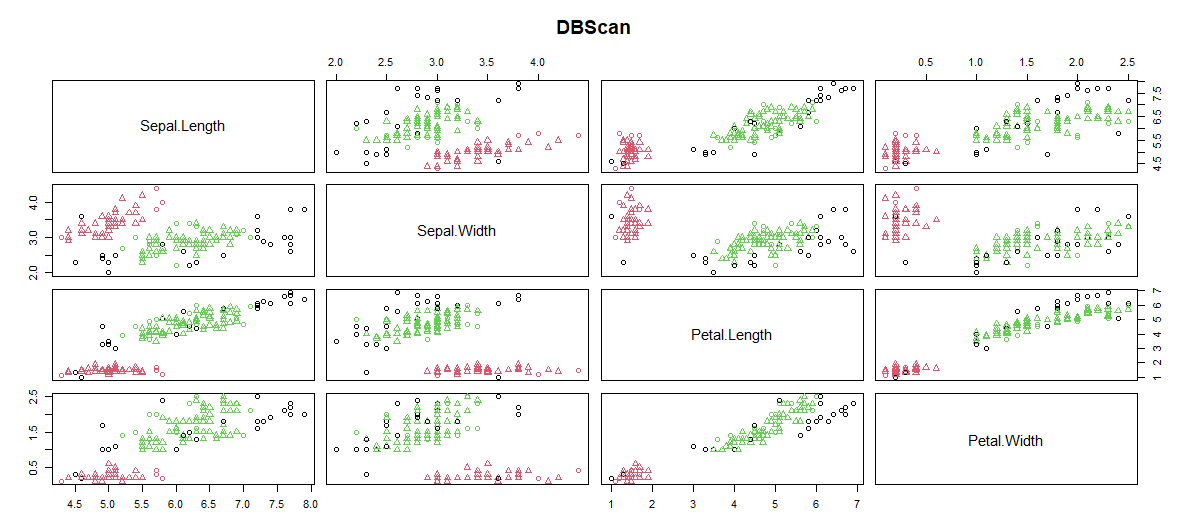
# 1. R语言dbscan聚类算法概述
聚类分析是数据挖掘中的一项关键技术,它将相似的对象分组在一起,以揭示数据的潜在结构。R语言作为数据科学领域常用的语言之一,提供了多种聚类算法实现,其中dbscan算法以其在处理任意形状的簇和噪声数据的能力而备受青睐。本章将概述dbscan算法的工作原理及其在R语言中的应用基础。
dbscan(Density-Based Spatial Clustering of Applications with Noise)是一种基于密度的空间聚类方法,它将具有足够高密度的区域划分为簇,并能在带有噪声的空间数据库中发现任意形状的聚类。与传统的k-means聚类算法相比,dbscan不需要预先指定簇的数量,这极大地简化了聚类过程。
R语言中的`dbscan`包提供了dbscan聚类算法的实现,它通过以下两个参数来控制聚类行为:Eps(邻域大小)和MinPts(形成簇所需的最小点数)。通过调整这两个参数,用户可以控制聚类的粒度,对数据集进行有效的聚类分析。接下来的章节,我们将深入探讨这两个参数的选择及其对聚类结果的影响。
```R
# R语言中使用dbscan算法的基本代码示例
library(dbscan)
# 假设data是一个R中的数据框或者矩阵
# eps和minPts是根据数据特征事先设定的参数
dbscan_result <- dbscan(data, eps = 0.5, minPts = 5)
```
在上述代码中,`dbscan`函数接受数据集和两个参数,返回聚类结果,其中聚类标号为正数的点被认为是核心对象,而标号为0的点是噪声。
# 2. dbscan聚类核心技巧
在深入探讨dbscan聚类核心技巧之前,有必要回顾一下该算法的基本原理。dbscan(Density-Based Spatial Clustering of Applications with Noise)是一种基于密度的空间聚类方法,它将具有足够高密度的区域划分为簇,并能在噪声中发现任意形状的聚类。
## 2.1 距离度量和邻域参数选择
dbscan算法中两个关键参数是Eps(邻域半径)和MinPts(核心点的最小邻居数),它们的合理选择对于聚类结果的质量至关重要。
### 2.1.1 Eps参数对聚类结果的影响
Eps参数定义了点的邻域大小,即用来决定点A和点B是否邻近的距离阈值。若点A的邻域内包含至少MinPts个点,则该点被认为是核心点。Eps设置过大,会导致不同的簇变得过于接近,甚至出现合并;Eps设置过小,则可能会造成簇被分割成多个部分。
**参数选择策略**:
- 选择Eps时可以使用k距离图(k-distance plot)。在该图中,横坐标是点按距离排序的索引,纵坐标是对应点到其第k近邻的距离。理想情况下,图中会有一处明显的“拐点”,该点之后的距离开始迅速增加,拐点对应的横坐标值可以作为Eps的参考值。
- 另一种方法是使用经验公式,结合数据集的特性,尝试多种Eps值,然后观察聚类结果的合理性。
### 2.1.2 MinPts参数的作用与选择
MinPts参数决定了形成一个簇所需的最少核心点数量。它与数据集的维度有关,维度越高,需要的最小核心点数通常越多。
**参数选择策略**:
- 一般而言,MinPts的取值应该大于等于数据集的维度加一,即MinPts ≥ dim(data) + 1。
- 对于高维数据,MinPts的取值可以更大,以便于识别出有意义的簇。但也不能太大,否则可能会导致所有数据点都被归为噪声。
## 2.2 高维数据的处理
高维数据集在许多领域中都是常见的,例如文本挖掘、生物信息学等。高维数据处理对于dbscan聚类至关重要,因为距离度量在高维空间中的表现与低维空间有很大差异,这被称为“维度的诅咒”。
### 2.2.1 高维数据的挑战与降维技术
- **挑战**:在高维空间中,数据点之间的距离可能变得非常相似,导致无法有效区分。这就是所谓的“距离集中效应”(distance concentration effect),会使得基于距离的聚类算法失效。
- **降维技术**:为了克服这个挑战,常用的技术包括主成分分析(PCA)、线性判别分析(LDA)等。这些技术可以减少数据集的维度,同时尽可能保留重要信息。
### 2.2.2 使用PCA等技术预处理高维数据
- **主成分分析(PCA)**:PCA是一种常用的数据降维技术,它通过正交变换将可能相关的高维变量转换为线性不相关的低维变量集,即主成分。前几个主成分包含了大部分数据变化的信息,因此可以用来代表原始数据集。
- **应用PCA步骤**:
1. 计算数据集的协方差矩阵。
2. 计算协方差矩阵的特征值和特征向量。
3. 将特征向量按对应特征值的大小排序,选择前k个最大的特征向量作为新的特征空间的基。
4. 将原始数据投影到这个新的k维特征空间中。
## 2.3 稀疏数据集的dbscan聚类
dbscan算法在稀疏数据集上的应用同样是一个挑战,因为稀疏性会导致算法难以识别出密度足够高的区域,进而影响聚类的效果。
### 2.3.1 稀疏矩阵简介
- **定义**:稀疏矩阵是一种矩阵,其中大部分元素的值为零。在数据分析中,稀疏矩阵的使用非常普遍,尤其是在文本处理和推荐系统中。
- **数据结构**:通常,稀疏矩阵可以使用特定的数据结构(如R语言中的`Matrix`包)来有效地存储和操作,只存储非零元素,从而节省内存。
### 2.3.2 在稀疏数据集上应用dbscan
- **优化策略**:对于稀疏数据集,可以通过修改距离计算方法来避免显式地计算所有元素之间的距离,以提高聚类效率。例如,可以使用投影技术将数据点映射到更低维度,然后在投影后的空间中应用dbscan。
- **示例代码块**:
```R
library("dbscan")
# 构建一个稀疏矩阵(这里仅为示例,实际应加载真实数据)
sparse_matrix <- Matrix::rsparsematrix(1000, 20, 0.05)
# 对稀疏矩阵进行聚类,选择一个合适的邻域半径Eps和核心点最小邻居数MinPts
clusters <- dbscan(sparse_matrix, eps=0.5, minPts=5)
```
在上述代码中,`rsparsematrix`函数生成了一个稀疏矩阵,`dbscan`函数用于在该稀疏矩阵上进行聚类。注意,由于稀疏矩阵的特殊性,Eps的值需要根据数据集的稀疏程度进行调整。
- **代码逻辑解读**:在R语言中,`Matrix`包提供了对稀疏矩阵的操作支持。通过`dbscan`函数,我们可以对稀疏矩阵进行聚类分析。需要特别注意的是,稀疏数据集的聚类参数Eps的选择,往往需要比普通数据集更细致的调整,以适应数据的稀疏特性。
通过以上内容,我们已经了解了如何选择合适的参数以及如何处理高维和稀疏数据集,以优化dbscan算法的性能。接下来的章节将讨论如何将这些技巧应用于实际数据集的聚类分析。
# 3. dbscan聚类实践应用
## 实际数据集的聚类分析
### 数据预处理步骤
在进行任何聚类分析之前,确保数据的质量至关重要。数据预处理的目的是为了确保输入数据对聚类算法友好,能够最大化其性能。以下是几个预处理数据时需要考虑的步骤:
1. **数据清洗**:
清除数据中的噪声和异常值,它们会干扰聚类结果,导致分群不准确。可使用统计方法识别并处理这些数据点,如 Z-score 标准化、IQR(四分位距)方法等。
2. **数据规范化**:
根据具体应用场景,可能需要对数据进行标准化或归一化处理。例如,标准化将数据按其均值和标准差进行转换,而归一化则将数据缩放到一个特定的范围,如 0 到 1。
3. **特征选择**:
选择合适的特征对于聚类分析非常重要,特征太少可能导致模型丢失关键信息,而太多则可能引入不相关或冗余的信息。可以使用相关性分析、主成分分析(PCA)等方法来辅助特征选择。
4. **数据转换**:
对于非数值型数据,需要将其转换为数值型,便于算法处理。常见的方法包括独热编码(One-Hot Encoding)或标签编码(Label Encoding)。
### 聚类过程详解与参数调整
在预处理数据之后,可以开始使用dbscan算法进行聚类。关键的参数调整步骤如下:
1. **Eps参数的调整**:
Eps是一个影响邻域大小的关键参数,影响着点之间的“密度可达”关系。需要根据数据的特性进行调整,通常使用KNN(K-最近邻)算法来帮助选取合适的Eps值。
2. **MinPts参数的调整**:
MinPts决定了形成一个聚类所需的最小点数。它与Eps一起决定了数据点被判定为核心点或边界点的条件。根据数据集的密度,需要通过试验来找到最合适的值。
3. **聚类实验**:
使用调整好的参数在数据集上运行dbscan算法,得到初步的聚类结果。然后根据聚类结果的分布和业务目标,对Eps和MinPts进行微调。
4. **结果验证与迭代**:
每次参数调整后,都需要重新评估聚类结果。可以使用轮廓系数(Silhouette Coefficient)等指标来衡量聚类效果,并进行迭代优化。
## 可视化展示聚类结果
### 使用ggplot2绘制聚类图形
聚类结果的可视化对于理解数据的结构至关重要。在R语言中,ggplot2是一个非常强大的可视化工具,可以用来展示聚类图形。以下是一个简单的示例代码,展示如何使用ggplot2来绘制dbscan聚类的结果:
```R
library(ggplot2)
# 假设已经有了一个数据框df,包含了聚类结果
df <- data.frame(
x = c(1, 2, 3, 4, 5, 6),
y = c(2, 1, 2, 3, 2, 1),
cluster = c(1, 1, 2, 2, 3, 3)
)
ggplot(df, aes(x=x, y=y, color=factor(cluster))) +
geom_point() +
labs(title="dbscan聚类结果可视化")
```
### 可视化中的高级技巧
当数据集具有多个维度时,使用ggplot2进行可视化可能会有挑战。这时可以采用一些高级技巧,如使用PCA对数据进行降维,然后绘制两维或三维的散点图来展示聚类效果。
## 聚类结果的评估与优化
### 聚类有效性指标的应用
聚类有效性指标用于评价聚类结果的质量,帮助我们判断聚类的合理性和有效性。以下是几种常用的聚类评估指标:
1. **轮廓系数**:
轮廓系数结合了聚类的凝聚度和分离度。它的取值范围在-1到1之间,值越大表示聚类效果越好。
2. **戴维森堡丁指数**(Davies-Bouldin Index):
这个指数是基于类内距离和类间距离的比率计算的,值越小表示聚类效果越好。
3. **Calinski-Harabasz指数**:
它是基于类间离散度和类内离散度的比率,指数高则聚类效果好。
### 根据评估结果调整参数
在评估聚类结果后,可能需要回到聚类过程中的参数调整步骤,重新调整Eps和MinPts参数。调整后,再次执行聚类和评估,形成一个迭代优化的过程。
这个过程可能需要重复多次,直到找到一个令人满意的聚类结果。实践中,这个迭代过程往往需要结合领域知识和业务理解,才能达到最佳的聚类效果。
以上所描述的内容是dbscan聚类实践应用的详细步骤和策略,为读者提供了一套完整的流程去理解和应用dbscan算法进行数据聚类分析。在下一章节中,我们将进一步探讨dbscan聚类的高级应用,以及如何与其他聚类算法结合使用来提高聚类效果。
# 4. dbscan聚类高级应用
## 4.1 处理不同规模的数据集
### 4.1.1 针对大数据集的优化技术
处理大数据集时,原始的dbscan算法可能会遇到性能瓶颈,因为算法需要计算大量点对之间的距离。大数据集中的点对计算是时间复杂度的主要来源。优化技术可以包括但不限于使用空间索引结构,如kd树、R树等,来减少必须考虑的点对数量。在R中,可以使用`dbscan`包的`FRNN`(Fast RNN)算法,这是一种基于空间索引的优化算法。例如,使用`dbscan::frNN()`函数可以提高大规模数据集的聚类速度。
```r
library(dbscan)
# 假设已经有一个大数据集:big_data
# 使用FRNN优化聚类
dbscan_result <- dbscan::frNN(big_data, eps = 0.5, minPts = 10)
# 输出聚类结果
print(dbscan_result)
```
在上述代码中,我们没有直接使用`dbscan`函数,而是使用了`frNN`函数,它通过构建近邻图来快速找到每个点的邻居,从而加速了聚类过程。`eps`和`minPts`是dbscan算法中的核心参数,需要根据数据集的特性进行适当调整。
### 4.1.2 小数据集的dbscan特有技巧
对于小型数据集,dbscan的表现通常很好,但如果数据集非常小,某些技巧可以帮助改善聚类质量。一个技巧是使用更多的邻域点来构建初始核心对象,这样可以增加密度连接点的数量,避免将本应属于同一群集的点错误地判定为噪声。此外,调整`eps`参数使之更小,以确保点群集之间有清晰的区分边界。
```r
# 对小型数据集进行聚类
dbscan_result_small <- dbscan(small_data, eps = 0.3, minPts = 5)
# 输出聚类结果
print(dbscan_result_small)
```
在此代码块中,`small_data`是小型数据集的变量名,`eps`被设置为0.3,`minPts`为5。由于数据集较小,我们可以选择较小的`eps`值来确保聚类结果的准确性。这种情况下,参数的选择更依赖于对数据的直观理解。
## 4.2 结合其他聚类算法进行分析
### 4.2.1 聚类算法的比较与结合
dbscan算法在发现任意形状的簇方面表现出色,但其计算成本较高,对于大数据集可能不够高效。因此,有时候我们可以考虑将dbscan与其他聚类算法结合使用,如先用k-means对数据进行初步聚类,然后再对结果应用dbscan以获得更精细的簇。这可以利用k-means的快速性能和dbscan的灵活性。
```r
# 使用k-means先进行初步聚类
kmeans_result <- kmeans(small_data, centers = 3)
# 将k-means的聚类结果用作dbscan的起始点
initial_points <- small_data[kmeans_result$cluster == 1,]
dbscan_result_combined <- dbscan(small_data, eps = 0.5, minPts = 5, start = initial_points)
# 输出最终聚类结果
print(dbscan_result_combined)
```
在这里,我们首先使用`kmeans`对数据集`small_data`进行聚类,假设我们想要识别3个簇。然后,我们选择其中一个簇的点作为dbscan的初始核心点(`start`参数),并执行dbscan聚类。这样可以利用k-means的效率和dbscan的灵活性,以期望得到更好的聚类结果。
### 4.2.2 实例:dbscan与k-means的对比应用
在实际应用中,我们可以将dbscan和k-means应用于相同的数据集,并对比其聚类结果。通过对比,我们可以了解不同算法的优缺点,并根据实际需求选择合适的算法。
| 聚类算法 | 优点 | 缺点 |
| --- | --- | --- |
| **dbscan** | 可以发现任意形状的簇;不需要预先指定簇的数量 | 计算成本较高;对噪声和离群点敏感 |
| **k-means** | 算法简单,速度快 | 需要预先指定簇的数量;只能发现凸形簇 |
## 4.3 算法的并行化和优化
### 4.3.1 并行计算的基本概念
并行计算是提高数据处理效率的关键技术之一,它通过同时使用多个计算资源来加速计算过程。在dbscan聚类算法中,可以将点对距离计算任务分配给多个处理器核心,以达到并行化处理的目的。在R中,可以使用`parallel`包提供的函数来实现简单的并行计算。
### 4.3.2 在R中实现dbscan的并行化处理
为了在R中实现dbscan的并行化处理,我们可以定义一个并行化的距离计算函数,并结合`mclapply`或`clusterApply`函数来并行执行计算。
```r
# 定义一个并行化的距离计算函数
par_dist <- function(data) {
library(parallel)
# 设定并行的核数
no_of_cores <- detectCores() - 1
# 使用clusterEvalQ来初始化并行计算环境
cluster <- makeCluster(no_of_cores)
clusterEvalQ(cluster, library(dbscan))
# 使用clusterApply进行并行计算
dists <- clusterApply(cluster, data, function(x) {
dist(x)
})
stopCluster(cluster)
# 返回计算结果的列表
dists
}
# 使用并行计算的距离进行dbscan聚类
dist_list <- par_dist(big_data)
dbscan_result_parallel <- dbscan(big_data, eps = 0.5, minPts = 10, dist = dist_list)
print(dbscan_result_parallel)
```
在这个例子中,我们首先定义了`par_dist`函数来计算数据点之间的距离矩阵。使用`makeCluster`创建一个并行计算集群,然后使用`clusterApply`将距离计算分配到集群中的每个核心。最后,我们使用`stopCluster`来关闭集群并释放资源。通过这种方式,我们实现了对大数据集的距离计算和聚类过程的并行化处理,显著提高了算法的运行效率。
# 5. dbscan聚类相关案例研究
本章将通过三个案例研究深入探讨dbscan聚类在不同领域的应用,通过每个案例的详细剖析,展示dbscan聚类算法的实际效用和分析方法。
## 5.1 生物信息学中的聚类分析
生物信息学是dbscan聚类应用的一个重要领域,尤其在基因表达数据和蛋白质结构预测中发挥着关键作用。
### 5.1.1 基因表达数据的聚类
基因表达数据通常包括成千上万个基因在不同样本中的表达水平。使用dbscan聚类可以识别出具有相似表达模式的基因群,这对于理解特定生物过程和疾病机理至关重要。
在R中,可以使用`dbscan`包来对基因表达矩阵进行聚类:
```R
library(dbscan)
gene_expression_data <- read.csv("gene_expression.csv")
dbscan_result <- dbscan(gene_expression_data, eps = 0.5, MinPts = 10)
plot(dbscan_result, data = gene_expression_data)
```
在上述代码中,`eps`和`MinPts`是dbscan算法的关键参数,需要根据具体的数据集进行调整。
### 5.1.2 聚类在蛋白质结构预测中的应用
蛋白质的三维结构决定了其功能,而聚类分析可以帮助识别具有相似结构特征的蛋白质群集。这在药物设计和功能预测中非常有用。
## 5.2 社交网络分析中的聚类应用
社交网络分析能够揭示社交群体的结构和模式,而dbscan聚类在其中扮演了识别群体和异常节点的角色。
### 5.2.1 社交网络数据的预处理
社交网络数据往往包含用户的社交关系和互动信息,预处理过程可能包括数据清洗、特征提取等。
在预处理后,可以使用dbscan聚类识别社交群体:
```R
social_network_data <- read.csv("social_network_data.csv")
dbscan_result <- dbscan(social_network_data, eps = 0.6, MinPts = 30)
plot(dbscan_result, data = social_network_data)
```
在这个案例中,聚类可以帮助我们发现紧密连接的社交群体,如兴趣小组或朋友网络。
### 5.2.2 检测社交群体的案例分析
通过dbscan聚类,我们可以进一步分析这些群体的特征。例如,可以使用R的`ggplot2`包来可视化社交群体的分布情况:
```R
library(ggplot2)
ggplot(social_network_data, aes(x, y, color = factor(dbscan_result$cluster))) +
geom_point() +
theme_minimal()
```
在这个图形中,不同的颜色代表不同的社交群体,可视化帮助我们直观地看到群体的形成。
## 5.3 市场细分中的聚类应用
市场细分利用聚类技术识别出具有相似特征和需求的消费者群体,这对制定有效的营销策略至关重要。
### 5.3.1 市场数据的聚类分析
市场数据可能包括消费者年龄、购买频率、收入水平等因素,dbscan聚类可以揭示消费者行为的自然分组。
使用dbscan进行聚类分析的代码示例如下:
```R
market_data <- read.csv("market_data.csv")
dbscan_result <- dbscan(market_data, eps = 0.4, MinPts = 20)
plot(dbscan_result, data = market_data)
```
根据聚类结果,企业可以将营销信息和产品定位到具体的消费者群体中。
### 5.3.2 从聚类结果到营销策略的转变
聚类结果可以指导营销策略的制定,例如,针对收入水平较高的群体,企业可能推出高端产品和服务。
通过dbscan聚类,公司能够更精细地理解其市场细分,并通过定制化的营销策略来增强竞争力。
在本章中,我们深入探讨了dbscan聚类在不同领域的具体应用,旨在展示其广泛的应用价值和在实际分析中的重要性。通过对生物信息学、社交网络分析和市场细分案例的研究,我们可以看到dbscan算法如何帮助我们揭示隐藏在数据中的模式和结构。这些案例不仅验证了dbscan聚类方法的适用性,而且为其他领域的应用提供了实际参考和灵感。

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏深入探讨了 R 语言中的 dbscan 数据包,提供了一系列详细教程和高级应用。涵盖了 dbscan 聚类的核心技巧、算法原理、参数调优、大数据处理、并行处理、非球形数据聚类、数据可视化、社交网络群体发现、图像分割等多个方面。通过深入浅出的讲解和丰富的案例研究,本专栏旨在帮助读者从新手到专家,全面掌握 dbscan 包的应用,提升聚类分析性能,解决聚类难题,并探索其在数据科学和机器学习领域的广泛应用。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【16位加法器设计秘籍】:全面揭秘高性能计算单元的构建与优化

# 摘要
本文对16位加法器进行了全面的研究和分析。首先回顾了加法器的基础知识,然后深入探讨了16位加法器的设计原理,包括二进制加法基础、组成部分及其高性能设计考量。接着,文章详细阐述
三菱FX3U PLC编程:从入门到高级应用的17个关键技巧

# 摘要
三菱FX3U PLC是工业自动化领域常用的控制器之一,本文全面介绍了其编程技巧和实践应用。文章首先概述了FX3U PLC的基本概念、功能和硬件结构,随后深入探讨了
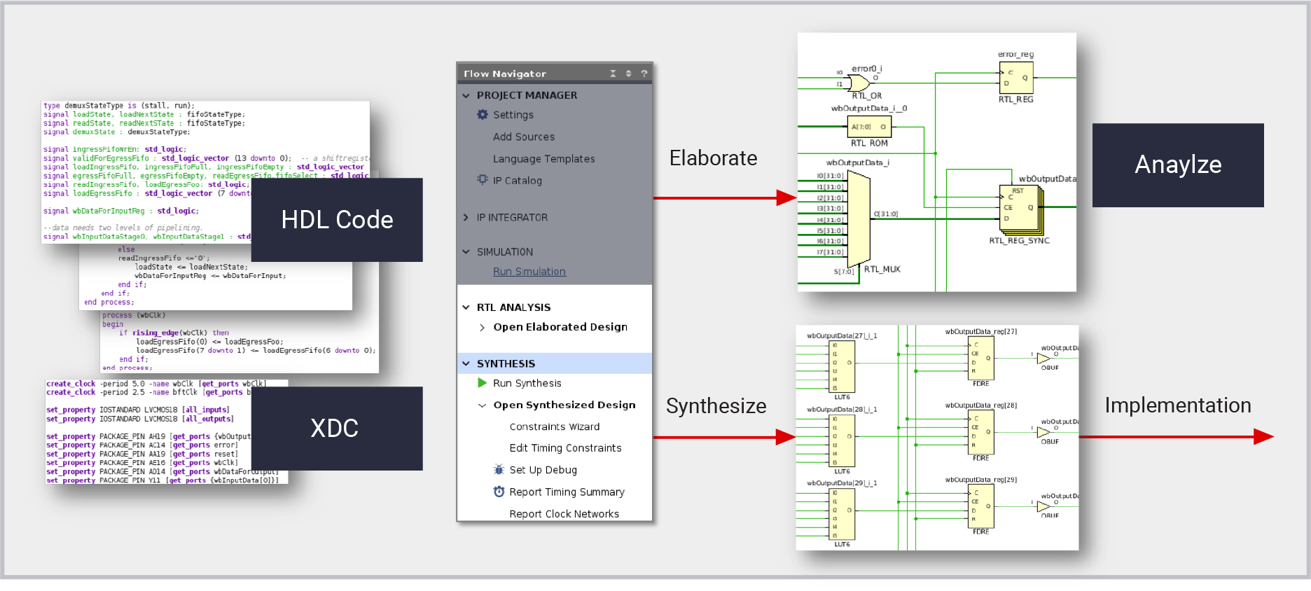
【Xilinx 7系列FPGA深入剖析】:掌握架构精髓与应用秘诀
# 摘要
本文详细介绍了Xilinx 7系列FPGA的关键特性及其在工业应用中的广泛应用。首先概述了7系列FPGA的基本架构,包括其核心的可编程逻辑单元(PL)、集成的块存储器(BRAM)和数字信号处理(DSP)单元。接着,本文探讨了使用Xilinx工具链进行FPGA编程与配置的流程,强调了设计优化和设备配置的重要性。文章进一步分析了7系列FPGA在
【图像技术的深度解析】:Canvas转JPEG透明度保护的终极策略

# 摘要
随着Web技术的不断发展,图像技术在前端开发中扮演着越来越重要的角色。本文首先介绍了图像技术的基础和Canvas绘
【MVC标准化:肌电信号处理的终极指南】:提升数据质量的10大关键步骤与工具
# 摘要
MVC标准化是肌电信号处理中确保数据质量的重要步骤,它对于提高测量结果的准确性和可重复性至关重要。本文首先介绍肌电信号的生理学原理和MVC标准化理论,阐述了数据质量的重要性及影响因素。随后,文章深入探讨了肌电信号预处理的各个环节,包括噪声识别与消除、信号放大与滤波技术、以及基线漂移的校正方法。在提升数据质量的关键步骤部分,本文详细描述了信号特征提取、MVC标准化的实施与评估,并讨论了数据质量评估与优化工具。最后,本文通过实验设计和案例分析,展示了MVC标准化在实践应用中的具
ISA88.01批量控制:电子制造流程优化的5大策略

# 摘要
本文首先概述了ISA88.01批量控制标准,接着深入探讨了电子制造流程的理论基础,包括原材料处理、制造单元和工作站的组成部分,以及流程控制的理论框架和优化的核心原则。进一步地,本文实
【Flutter验证码动画效果】:如何设计提升用户体验的交互

# 摘要
随着移动应用的普及和安全需求的提升,验证码动画作为提高用户体验和安全性的关键技术,正受到越来越多的关注。本文首先介绍Flutter框架下验证码动画的重要性和基本实现原理,涵盖了动画的类型、应用场景、设计原则以及开发工具和库。接着,文章通过实践篇深入探讨了在Flutter环境下如何具体实现验证码动画,包括基础动画的制作、进阶技巧和自定义组件的开发。优化篇
ENVI波谱分类算法:从理论到实践的完整指南
# 摘要
ENVI软件作为遥感数据处理的主流工具之一,提供了多种波谱分类算法用于遥感图像分析。本文首先概述了波谱分类的基本概念及其在遥感领域的重要性,然后介绍了ENVI软件界面和波谱数据预处理的流程。接着,详细探讨了ENVI软件中波谱分类算法的实现方法,通过实践案例演示了像元级和对象级波谱分类算法的操作。最后,文章针对波谱分类的高级应用、挑战及未来发展进行了讨论,重点分析了高光谱数据分类和深度学习在波谱分类中的应用情况,以及波谱分类在土地覆盖制图和农业监测中的实际应用。
# 关键字
ENVI软件;波谱分类;遥感图像;数据预处理;分类算法;高光谱数据
参考资源链接:[使用ENVI进行高光谱分
【天线性能提升密籍】:深入探究均匀线阵方向图设计原则及案例分析
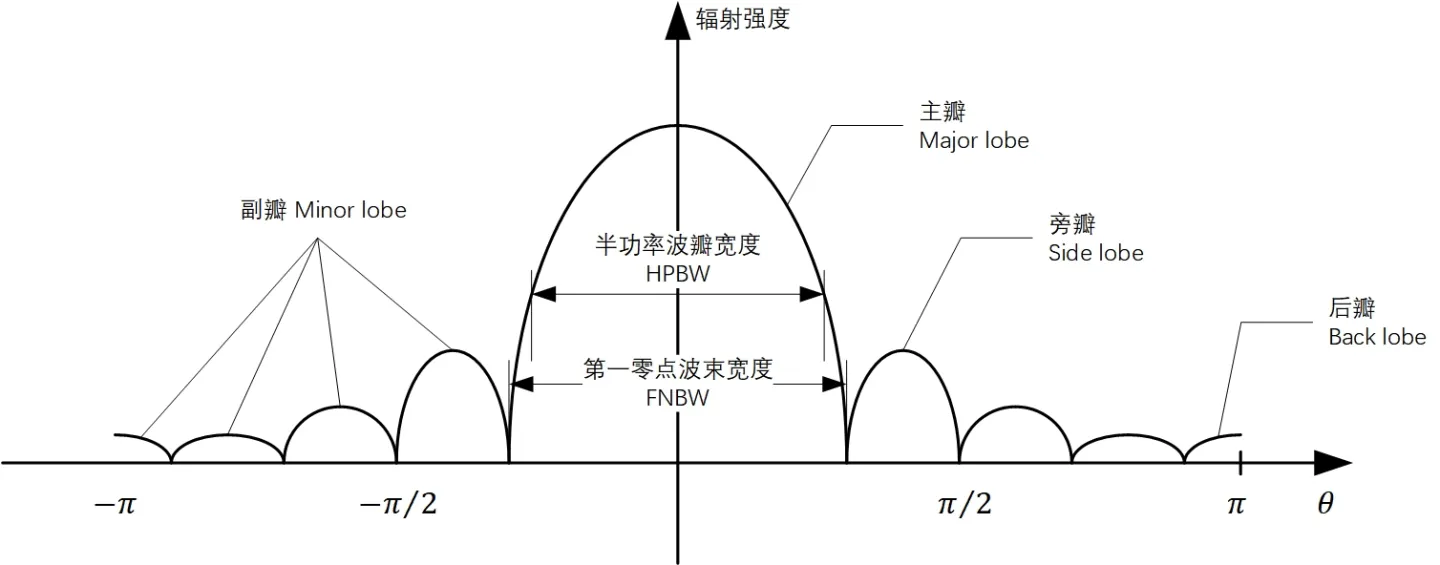
# 摘要
本文深入探讨了均匀线阵天线的基础理论及其方向图设计,旨在提升天线系统的性能和应用效能。文章首先介绍了均匀线阵及方向图的基本概念,并阐述了方向图设计的理论基础,包括波束形成与主瓣及副瓣特性的控制。随后,论文通过设计软件工具的应用和实际天线系统调试方法,展示了方向图设计的实践技巧。文中还包含了一系列案例分析,以实证研究验证理论,并探讨了均匀线阵性能
【兼容性问题】快解决:专家教你确保光盘在各设备流畅读取
/i.s3.glbimg.com/v1/AUTH_08fbf48bc0524877943fe86e43087e7a/internal_photos/bs/2021/L/w/I3DfXKTAmrqNi0rGtG5A/2014-06-24-cd-dvd-bluray.png)
# 摘要
光盘作为一种传统的数据存储介质,其兼容性问题长
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )