Seurat对象中的数据可视化技巧:展现单细胞数据的奥秘
发布时间: 2024-03-30 14:59:20 阅读量: 54 订阅数: 43 

Seurat-to-RNA-Velocity:将Seurat对象与RNA Velocity结合使用的指南
# 1. 理解Seurat对象
## 1.1 什么是Seurat对象?
在单细胞RNA测序数据分析中,Seurat对象是一个重要的数据结构,用于存储和管理单细胞数据的各种信息,包括基因表达矩阵、细胞特征数据、聚类结果、降维结果等。
## 1.2 Seurat对象在单细胞数据分析中的作用
Seurat对象可以帮助研究人员对单细胞数据进行处理、分析和可视化,从而揭示细胞类型、细胞亚群、基因表达模式等重要信息,为研究提供深入的见解。
## 1.3 如何创建和操作Seurat对象
在R语言中,可以使用Seurat包中的函数来创建和操作Seurat对象,包括导入数据、数据预处理、聚类分析、降维可视化等操作。熟练掌握Seurat对象的创建和操作是进行单细胞数据分析的基础。
# 2. 单细胞数据的预处理
在单细胞数据分析中,数据的预处理是非常重要的一步,它包括数据的质控、降噪、标准化、归一化以及细胞类型鉴定与聚类分析等内容。下面我们将详细介绍单细胞数据的预处理过程。
### 2.1 数据质控与降噪
在数据质控阶段,我们需要对数据进行初步的筛选,去除低质量的细胞和异常数据点,以保证后续分析的准确性。常用的数据质控指标包括细胞的基因表达数目、细胞的mapping率等。
接下来是降噪步骤,降噪的目的是去除数据中的噪声点,提高数据的质量。在单细胞数据中,常用的降噪方法包括使用PCA、ICA等降维技术,以及一些专门针对单细胞数据的降噪算法,如scVI、SAVER等。
### 2.2 数据标准化与归一化
数据标准化是为了消除不同细胞之间的技术差异,使得不同细胞之间的基因表达值具有可比性。常见的标准化方法包括Z-score标准化、Log转换等。
数据归一化则是为了消除不同基因之间的表达量差异,使得各个基因在后续分析中具有相同的权重。归一化方法有TPM归一化、RPKM归一化等。
### 2.3 细胞类型鉴定与聚类分析
在数据预处理的最后阶段,我们需要对细胞进行类型鉴定和聚类分析,将相似的细胞聚在一起形成细胞亚群。常用的聚类方法包括K-means、DBSCAN、Hierarchical clustering等。经过聚类分析,我们可以更好地了解数据中不同细胞类型的分布情况,为后续的细胞亚群分析奠定基础。
通过以上步骤,我们可以对单细胞数据进行有效的预处理,为后续的分析和可视化工作打下坚实的基础。
# 3. 应用PCA和t-SNE进行降维可视化
在单细胞数据分析中,降维可视化是一种常见且有效的手段,可以帮助我们更好地理解数据的结构和特征。本章将介绍如何利用主成分分析(PCA)和 t-分布邻域嵌入(t-SNE)这两种降维算法对单细胞数据进行可视化。
#### 3.1 什么是PCA和t-SNE?
- **PCA(Principal Component Analysis)**:主成分分析是一种常用的线性降维技术,它通过将原始数据投影到新的正交坐标轴上,以发现数据中最重要的模式和结构。PCA寻找数据中的主成分(即方差最大的方向),从而减少数据的维度。
- **t-SNE(t-Distributed Stochastic Neighbor Embedding)**:t-SNE是一种非线性的降维算法,它可以将高维数据映射到二维或三维空间,并保留数据点之间的局部结构。t-SNE在可视化高维数据方面效果非常好,特别适用于发现数据中的聚类和簇状结构。
#### 3.2 如何利用PCA和t-SNE对单细胞数据进行降维
在Seurat中,对单细胞数据进行PCA和t-SNE降维可视化非常简单。下面是一个基本的代码示例:
```python
# 运行PCA
pbmc <- ScaleData(pbmc, features = rownames(pbmc))
pbmc <- RunPCA(pbmc, npcs = 30)
pbmc <- ProjectPCA(pbmc, do.center =
```

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏将全面介绍Seurat对象在单细胞RNA分析中的应用。从初识Seurat对象的核心数据结构,到基础操作指南的轻松上手,再到高级功能解析的数据处理与可视化,以及数据预处理、细胞聚类算法、细胞亚群鉴定、基因表达分析、细胞间相互作用分析、时间序列分析等方面深入探讨。此外,探讨Seurat对象在免疫细胞研究、疾病研究、药物筛选、细胞类型识别、多组学数据集成、功能富集分析等领域的应用,揭示Seurat对象在创新医学研究中的关键角色。通过本专栏,读者将深入了解Seurat对象在单细胞数据分析中的多方面应用,为探索新颖医学发现提供数据支撑。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
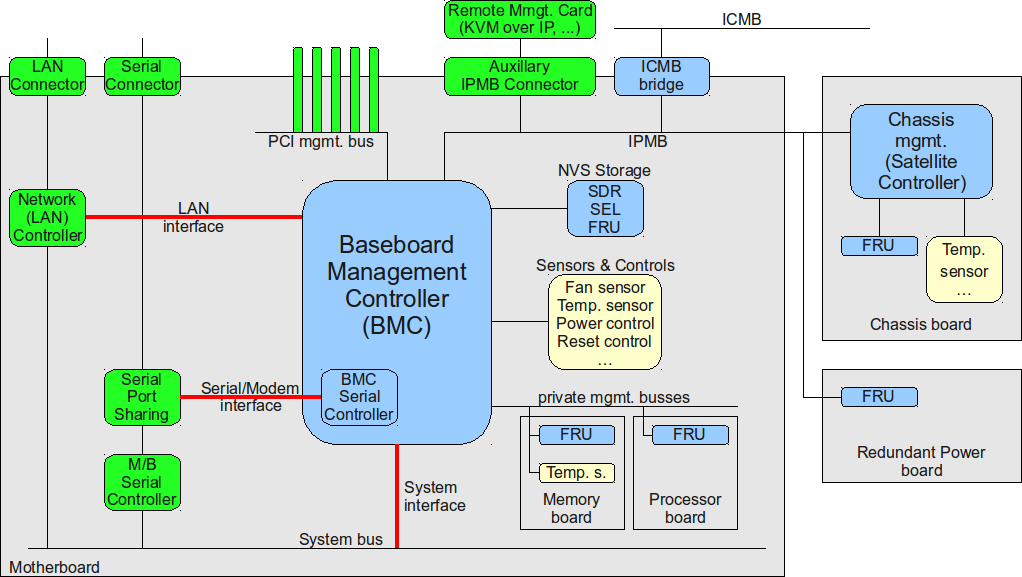
IPMI标准V2.0与物联网:实现智能设备自我诊断的五把钥匙
# 摘要
本文旨在深入探讨IPMI标准V2.0在现代智能设备中的应用及其在物联网环境下的发展。首先概述了IPMI标准V2.0的基本架构和核心理论,重点分析了其安全机制和功能扩展。随后,本文讨论了物联网设备自我诊断的必要性,并展示了IPMI标准V2.0在智能硬件设备和数据中心健康管理中的应用实例。最后,本文提出了实现智能设备IPMI监控系统的设计与开发指南,
【EDID兼容性高级攻略】:跨平台显示一致性的秘诀

# 摘要
电子显示识别数据(EDID)是数字视频接口中用于描述显示设备特性的标准数据格式。本文全面介绍了EDID的基本知识、数据结构以及兼容性问题的诊断与解决方法,重点关注了数据的深度解析、获取和解析技术。同时,本文探讨了跨平台环境下EDID兼容性管理和未来技术的发展趋势,包括增强型EDID标准的发展和自动化配置工具的前景。通过案例研究与专家建议,文章提供了在多显示器设置和企业级显示管理中遇到的ED
PyTorch张量分解技巧:深度学习模型优化的黄金法则

# 摘要
PyTorch张量分解技巧在深度学习领域具有重要意义,本论文首先概述了张量分解的概念及其在深度学习中的作用,包括模型压缩、加速、数据结构理解及特征提取。接着,本文详细介绍了张量分解的基础理论,包括其数学原理和优化目标,随后探讨了在PyTorch中的操作实践,包括张量的创建、基本运算、分解实现以及性能评估。论文进一步深入分析了张量分解在深度学习模型中的应用实例,展示如何通过张量分解技术实现模型
【参数校准艺术】:LS-DYNA材料模型方法与案例深度分析
# 摘要
本文全面探讨了LS-DYNA软件在材料模型参数校准方面的基础知识、理论、实践方法及高级技术。首先介绍了材料模型与参数校准的基础知识,然后深入分析了参数校准的理论框架,包括理论与实验数据的关联以及数值方法的应用。文章接着通过实验准备、模拟过程和案例应用详细阐述了参数校准的实践方法。此外,还探
系统升级后的验证:案例分析揭秘MAC地址修改后的变化

# 摘要
本文系统地探讨了MAC地址的基础知识、修改原理、以及其对网络通信和系统安全性的影响。文中详细阐述了软件和硬件修改MAC地址的方法和原理,并讨论了系统升级对MAC地址可能产生的变化,包括自动重置和保持不变的情况。通过案例分析,本文进一步展示了修改MAC地址后进行系统升级的正反两面例子。最后,文章总结了当前研究,并对今后关于MAC地址的研究方向进行了展望。
# 关键字
华为交换机安全加固:5步设置Telnet访问权限
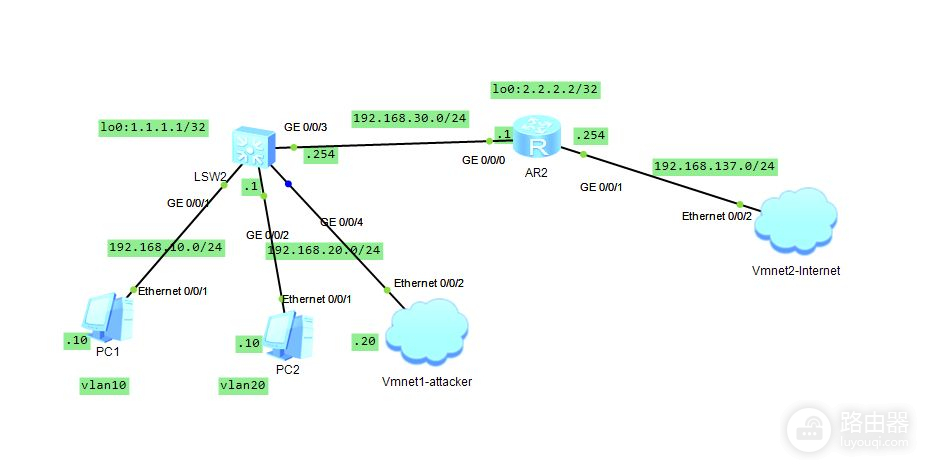
# 摘要
随着网络技术的发展,华为交换机在企业网络中的应用日益广泛,同时面临的安全威胁也愈加复杂。本文首先介绍了华为交换机的基础知识及其面临的安全威胁,然后深入探讨了Telnet协议在交换机中的应用以及交换机安全设置的基础知识,包括用户认证机制和网络接口安全。接下来,文章详细说明了如何通过访问控制列表(ACL)和用户访问控制配置来实现Telnet访问权限控制,以增强交换机的安全性。最后,通过具体案例分析,本文评估了安
【软硬件集成测试策略】:4步骤,提前发现并解决问题
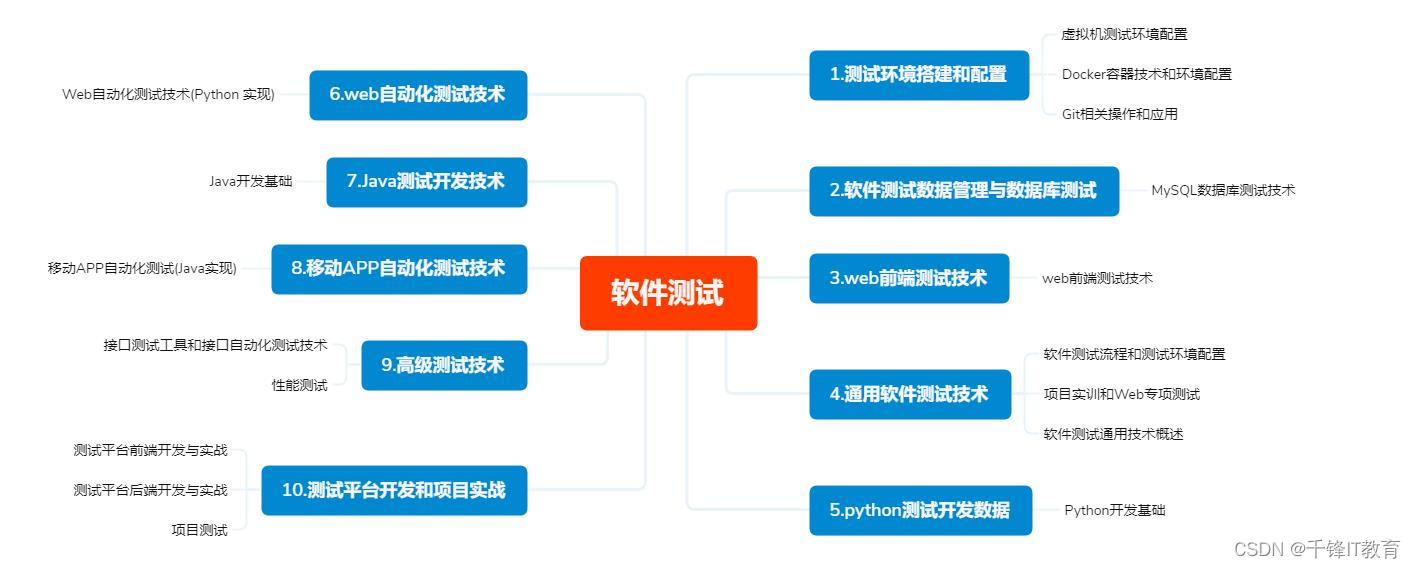
# 摘要
软硬件集成测试是确保产品质量和稳定性的重要环节,它面临诸多挑战,如不同类型和方法的选择、测试环境的搭建,以及在实践操作中对测试计划、用例设计、缺陷管理的精确执行。随着技术的进步,集成测试正朝着性能、兼容性和安全性测试的方向发展,并且不断优化测试流程和数据管理。未来趋势显示,自动化、人工智能和容器化等新兴技术的应用,将进一步提升测试效率和质量。本文系统地分析了集成测试的必要性、理论基础、实践操作
CM530变频器性能提升攻略:系统优化的5个关键技巧

# 摘要
本文综合介绍了CM530变频器在硬件与软件层面的优化技巧,并对其性能进行了评估。首先概述了CM530的基本功能与性能指标,然后深入探讨了硬件升级方案,包括关键硬件组件选择及成本效益分析,并提出了电路优化和散热管理的策略。在软件配置方面,文章讨论了软件更新流程、固件升级准备、参数调整及性能优化方法。系统维护与故障诊断部分提供了定期维护的策略和故障排除技巧。最后,通过实战案例分析,展示了CM530在特定应用中的优化效果,并对未来技术发展和创新
CMOS VLSI设计全攻略:从晶体管到集成电路的20年技术精华
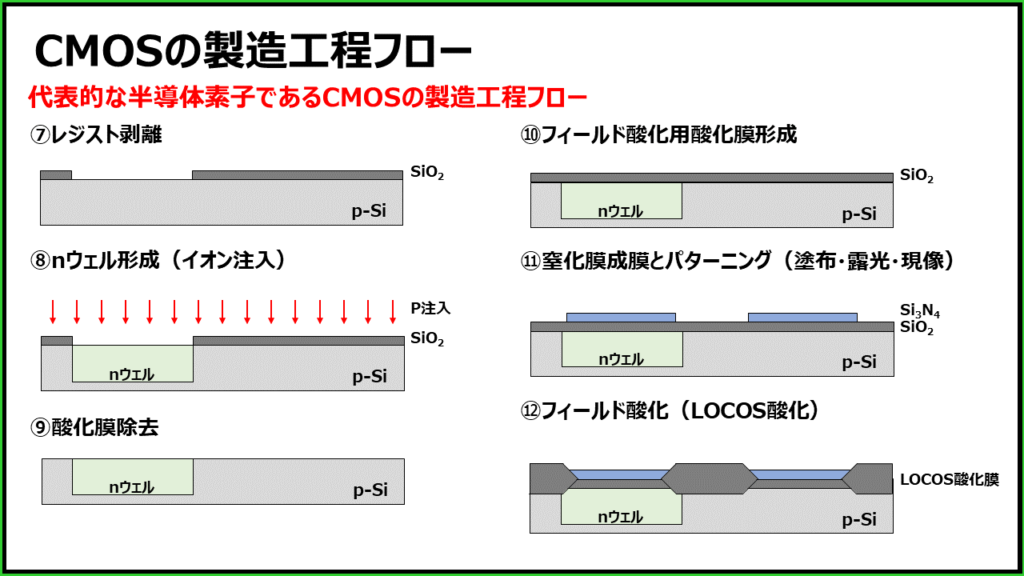
# 摘要
本文对CMOS VLSI设计进行了全面概述,从晶体管级设计基础开始,详细探讨了晶体管的工作原理、电路模型以及逻辑门设计。随后,深入分析了集成电路的布局原则、互连设计及其对信号完整性的影响。文章进一步介绍了高级CMOS电路技术,包括亚阈值电路设计、动态电路时序控制以及低功耗设计技术。最后,通过VLSI设计实践和案例分析,阐述了设计流程、
三菱PLC浮点数运算秘籍:精通技巧全解

# 摘要
本文系统地介绍了三菱PLC中浮点数运算的基础知识、理论知识、实践技巧、高级应用以及未来展望。首先,文章阐述了浮点数运算的基础和理论知识,包括表示方法、运算原理及特殊情况的处理。接着,深入探讨了三菱PLC浮点数指令集、程序设计实例以及调试与优化方法。在高级应用部分,文章分析了浮点数与变址寄存器的结合、高级算法应用和工程案例。最后,展望了三菱PLC浮点数运算技术的发展趋势,以及与物联网的结合和优化
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )