模式匹配在生物信息学中的应用
发布时间: 2023-12-20 12:16:07 阅读量: 48 订阅数: 27 
## 1. 序言
### 生物信息学简介
生物信息学是一门跨学科的科学,主要研究生物学、计算机科学和数学的交叉学科领域。其主要任务是通过生物信息的存储、检索和分析,揭示生物学内在规律。生物信息学的发展为生物学研究提供了强大的工具和方法。
### 模式匹配的基本概念
模式匹配是生物信息学中常用的一种方法,它能够在生物序列数据中寻找特定的模式或序列。模式匹配可以帮助研究者发现DNA、RNA或蛋白质序列中的重要特征,如基因、启动子、蛋白质结构等。在生物信息学中,模式匹配算法的应用十分广泛,对于深入理解生物学系统和进行生物学研究具有重要意义。
## 2. DNA序列分析中的模式匹配
DNA序列是生物体遗传信息的载体,对DNA序列进行分析可以帮助科学家们理解生物体的遗传特征和变化规律。模式匹配在DNA序列分析中起着至关重要的作用,主要包括以下方面的应用:
### DNA序列的特点和序列比对
DNA序列通常由四种核苷酸(腺嘌呤,胸腺嘧啶,鸟嘌呤,胞嘧啶)组成,具有特定的序列特征和排列方式。通过模式匹配算法,可以对DNA序列进行比对和定位,从而找到相同或相似的序列片段,进行进化分析、物种分类等研究。
```python
# Python示例代码
import re
# 定义模式
pattern = "ATGCG"
# 定义DNA序列
dna_sequence = "ATGCGTACGCGTACGATCG"
# 进行模式匹配
matches = re.finditer(pattern, dna_sequence)
# 输出匹配结果
for match in matches:
print("找到匹配:", match.group(), " 位置:", match.start())
```
代码解释:
- 使用Python的re模块进行模式匹配
- 定义DNA序列的模式为"ATGCG"
- 通过finditer函数找到匹配的位置并输出
### 基因识别和启动子预测
模式匹配算法可以帮助科学家们识别DNA序列中的基因编码区域,进而预测基因的结构和功能。此外,还可以用于预测启动子和转录因子结合位点,揭示基因的调控机制。
```java
// Java示例代码
import java.util.regex.Matcher;
import java.util.regex.Pattern;
public class GenePrediction {
public static void main(String[] args) {
String pattern = "ATG(?:...)*?TAA";
String dnaSequence = "ATGCGTACGCGTACGATCGTAAGCTAGCTAGCTA";
Pattern p = Pattern.compile(pattern);
Matcher m = p.matcher(dnaSequence);
while (m.find()) {
System.out.println("找到匹配: " + m.group() + " 位置: " + m.start());
}
}
}
```
代码解释:
- 使用Java的正则表达式进行模式匹配
- 定义基因编码序列的模式为"ATG(?:...)*?TAA"
- 通过Matcher对象找到匹配的位置并输出
### 编程语言中模式匹配算法的应用
除了使用现成的模式匹配工具,编程语言本身也提供了丰富的模式匹配功能,如Python中的re模块、Java中的正则表达式等。通过编程语言的模式匹配功能,可以更灵活地处理DNA序列的分析和处理。
DNA序列分析中的模式匹配是生物信息学研究的重要内容之一,通过模式匹配算法,可以更好地理解DNA序列的结构和功能,推动生命科学领域的发展。
以上是DNA序列分析中模式匹配的部分应用,下一节我们将深入探讨模式匹配在蛋白质序列分析中的应用。
### 3. 蛋白质序列分析中的模式匹配
蛋白质是生物体内功能和结构的重要组成部分,其序列包含丰富的信息,利用模式匹配技术可以对蛋白质序列进行多方面的分析和预测。蛋白质序列分析中的模式匹配应用广泛,涉及蛋白质结构预测、蛋白质功能预测和蛋白质相互作用预测等多个领域。
1. **蛋白质结构预测**
蛋白质结构预测是生物信息学中的重要问题之一。模式匹配技术可以用于在已知蛋白质结构数据库中搜索相似的结构,从而推断待预测蛋白质的可能结构。在这个过程中,常用的模式匹配算法包括Smith-Waterman算法和BLAST算法,它们可以在蛋白质序列间找到局部和全局的相似度。
```python
# Python示例代码
from Bio import pairwise2
from Bio.pairwise2 import format_alignment
# 用Smith-Waterman算法比对两个蛋白质序列
X = "GATTACA"
Y = "GCATGCT"
alignments = pairwise2.align.localxx(X, Y)
for a in alignments:
print(format_alignment(*a))
```
以上代码演示了如何使用Biopython库进行Smith-Waterman算法的局部比对,从而预测蛋白质结构。
结果说明:该算法将序列"GATTACA"与"GCATGCT"进行比对,找到了它们之间的局部相似性,并输出了比对结果。
2. **蛋白质功能预测**
利用模式匹

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏旨在深入探讨模式匹配算法在各个领域中的应用与实践。从基本概念到高级技术,涵盖了字符串、文本、图像、音频等多种类型的模式匹配算法。文章包括了暴力匹配、KMP算法、正则表达式、通配符匹配、Boyer-Moore算法、AC自动机、Trie树等经典算法的详细解析,同时还介绍了Levenshtein距离、Jaccard相似性、余弦相似度等模糊匹配算法以及深度学习、机器学习在模式匹配中的应用。此外,还涵盖了模式匹配在自然语言处理、生物信息学、金融领域的具体应用案例。无论你是初学者还是专业人士,本专栏都将帮助你深入了解模式匹配算法的原理与实践,掌握多领域的模式匹配技术,为实际问题的解决提供有力支持。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
JY01A直流无刷IC全攻略:深入理解与高效应用
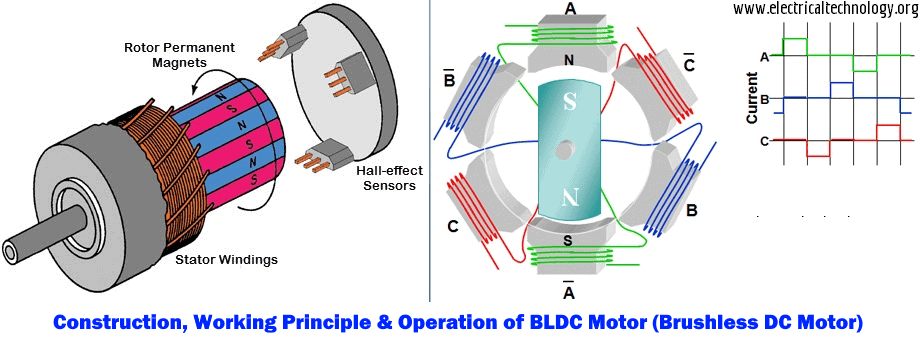
# 摘要
本文详细介绍了JY01A直流无刷IC的设计、功能和应用。文章首先概述了直流无刷电机的工作原理及其关键参数,随后探讨了JY01A IC的功能特点以及与电机集成的应用。在实践操作方面,本文讲解了JY01A IC的硬件连接、编程控制,并通过具体
【S参数转换表准确性】:实验验证与误差分析深度揭秘
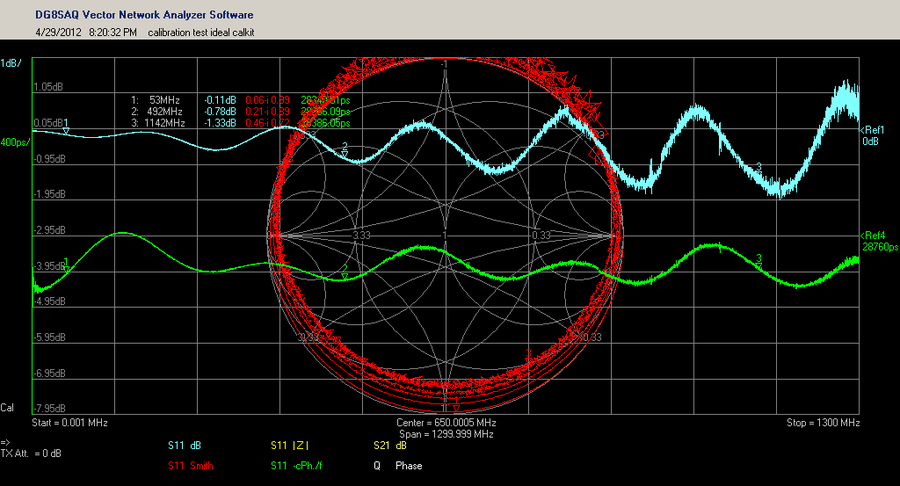
# 摘要
本文详细探讨了S参数转换表的准确性问题,首先介绍了S参数的基本概念及其在射频领域的应用,然后通过实验验证了S参数转换表的准确性,并分析了可能的误差来源,包括系统误差和随机误差。为了减小误差,本文提出了一系列的硬件优化措施和软件算法改进策略。最后,本文展望了S参数测量技术的新进展和未来的研究方向,指出了理论研究和实际应用创新的重要性。
# 关键字
S参
【TongWeb7内存管理教程】:避免内存泄漏与优化技巧
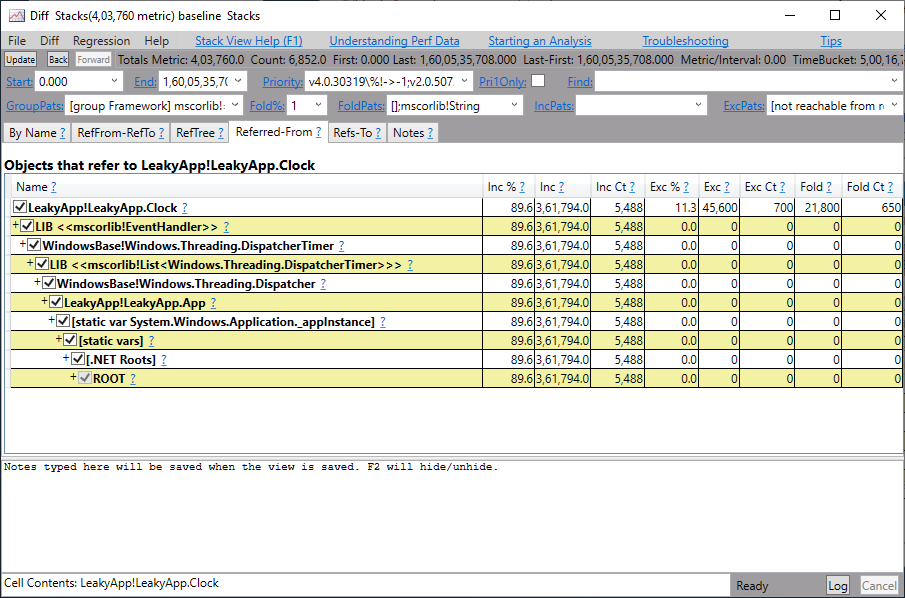
# 摘要
本文旨在深入探讨TongWeb7的内存管理机制,重点关注内存泄漏的理论基础、识别、诊断以及预防措施。通过详细阐述内存池管理、对象生命周期、分配释放策略和内存压缩回收技术,文章为提升内存使用效率和性能优化提供了实用的技术细节。此外,本文还介绍了一些性能优化的基本原则和监控分析工具的应用,以及探讨了企业级内存管理策略、自动内存管理工具和未来内存管理技术的发展趋
无线定位算法优化实战:提升速度与准确率的5大策略
# 摘要
本文综述了无线定位技术的原理、常用算法及其优化策略,并通过实际案例分析展示了定位系统的实施与优化。第一章为无线定位技术概述,介绍了无线定位技术的基础知识。第二章详细探讨了无线定位算法的分类、原理和常用算法,包括距离测量技术和具体定位算法如三角测量法、指纹定位法和卫星定位技术。第三章着重于提升定位准确率、加速定位速度和节省资源消耗的优化策略。第四章通过分析室内导航系统和物联网设备跟踪的实际应用场景,说明了定位系统优化实施
成本效益深度分析:ODU flex-G.7044网络投资回报率优化
# 摘要
本文旨在介绍ODU flex-G.7044网络技术及其成本效益分析。首先,概述了ODU flex-G.7044网络的基础架构和技术特点。随后,深入探讨成本效益理论,包括成本效益分析的基本概念、应用场景和局限性,以及投资回报率的计算与评估。在此基础上,对ODU flex-G.7044网络的成本效益进行了具体分析,考虑了直接成本、间接成本、潜在效益以及长期影响。接着,提出优化投资回报
【Delphi编程智慧】:进度条与异步操作的完美协调之道

# 摘要
本文旨在深入探讨Delphi编程环境中进度条的使用及其与异步操作的结合。首先,基础章节解释了进度条的工作原理和基础应用。随后,深入研究了Delphi中的异步编程机制,包括线程和任务管理、同步与异步操作的原理及异常处理。第三章结合实
C语言编程:构建高效的字符串处理函数
# 摘要
字符串处理是编程中不可或缺的基础技能,尤其在C语言中,正确的字符串管理对程序的稳定性和效率至关重要。本文从基础概念出发,详细介绍了C语言中字符串的定义、存储、常用操作函数以及内存管理的基本知识。在此基础上,进一步探讨了高级字符串处理技术,包括格式化字符串、算法优化和正则表达式的应用。
【抗干扰策略】:这些方法能极大提高PID控制系统的鲁棒性
# 摘要
PID控制系统作为一种广泛应用于工业过程控制的经典反馈控制策略,其理论基础、设计步骤、抗干扰技术和实践应用一直是控制工程领域的研究热点。本文从PID控制器的工作原理出发,系统介绍了比例(P)、积分(I)、微分(D)控制的作用,并探讨了系统建模、控制器参数整定及系统稳定性的分析方法。文章进一步分析了抗干扰技术,并通过案例分析展示了PID控制在工业温度和流量控制系统中的优化与仿真。最后,文章展望了PID控制系统的高级扩展,如
业务连续性的守护者:中控BS架构考勤系统的灾难恢复计划

# 摘要
本文旨在探讨中控BS架构考勤系统的业务连续性管理,概述了业务连续性的重要性及其灾难恢复策略的制定。首先介绍了业务连续性的基础概念,并对其在企业中的重要性进行了详细解析。随后,文章深入分析了灾难恢复计划的组成要素、风险评估与影响分析方法。重点阐述了中控BS架构在硬件冗余设计、数据备份与恢复机制以及应急响应等方面的策略。
自定义环形菜单

# 摘要
本文探讨了环形菜单的设计理念、理论基础、开发实践、测试优化以及创新应用。首先介绍了环形菜单的设计价值及其在用户交互中的应用。接着,阐述了环形菜单的数学基础、用户交互理论和设计原则,为深入理解环形菜单提供了坚实的理论支持。随后,文章详细描述了环形菜单的软件实现框架、核心功能编码以及界面与视觉设计的开发实践。针对功能测试和性能优化,本文讨论了测试方法和优化策略,确保环形菜单的可用性和高效性。最后,展望了环形菜单在新兴领域的
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )