DFT在材料科学中的应用:晶体结构分析与材料表征的必备工具
发布时间: 2024-07-02 14:05:43 阅读量: 162 订阅数: 54 

# 1. DFT在材料科学中的理论基础**
密度泛函理论(DFT)是一种强大的计算方法,用于研究材料的电子结构和性质。它基于这样一个假设:一个体系的总能量可以表示为电子密度的泛函。
DFT的数学基础是霍亨伯格-科恩定理,该定理指出,体系的总能量是电子密度的唯一泛函。这意味着,如果已知体系的电子密度,则可以完全确定其能量和所有其他性质。
DFT的实用性在于,它将多体薛定谔方程简化为一个求解电子密度的自洽方程组。通过使用近似的交换相关泛函来描述电子之间的相互作用,DFT能够在计算上可行的成本下提供准确的电子结构信息。
# 2. DFT计算方法与实践**
**2.1 DFT的数学原理和计算方法**
**2.1.1 密度泛函理论的基本原理**
密度泛函理论(DFT)是一种从头算的量子力学方法,用于计算多电子体系的电子结构和性质。DFT的基本原理是:一个体系的总能量可以表示为电子密度的泛函,即:
```
E[ρ] = T[ρ] + U[ρ] + Exc[ρ]
```
其中:
* E[ρ] 为体系的总能量
* T[ρ] 为体系的动能
* U[ρ] 为体系的电子间相互作用能
* Exc[ρ] 为体系的交换关联能
DFT通过近似交换关联能Exc[ρ]来简化计算。常用的交换关联泛函有:
* 局域密度近似(LDA)
* 广义梯度近似(GGA)
* 杂化泛函(如B3LYP)
**2.1.2 常用交换相关泛函及其选择**
选择合适的交换相关泛函对于DFT计算的精度至关重要。不同泛函适用于不同的体系和性质。
| 泛函类型 | 特点 | 适用范围 |
|---|---|---|
| LDA | 计算简单,精度较低 | 电子密度分布均匀的体系 |
| GGA | 计算精度较高,但计算量较大 | 电子密度分布不均匀的体系 |
| 杂化泛函 | 计算精度最高,但计算量最大 | 涉及共价键或激发态的体系 |
**2.2 DFT计算软件和实践应用**
**2.2.1 DFT软件的选用和安装**
常用的DFT软件包括:
* VASP
* Quantum Espresso
* Gaussian
* ADF
软件的选择取决于体系的大小、性质和计算需求。安装过程一般遵循软件文档的说明。
**2.2.2 DFT计算参数的设置和优化**
DFT计算参数包括:
* 基组:描述电子波函数的基函数集合
* 赝势:近似描述原子核和内层电子的作用
* 截断能:平面波基组的能量截断值
* k点:布里渊区的采样点
这些参数需要根据体系和计算精度进行优化。优化过程通常涉及多次计算和参数调整。
**代码块:VASP计算参数设置示例**
```
# VASP计算参数设置
INCAR = {
"ENCUT" : 520, # 截断能
"KPOINTS" : {
"MonkhorstPack" : [4, 4, 4], # k点采样
},
"EDIFF" : 1e-6, # 能量收敛阈值
"ISMEAR" : 0, # 电子占据态处理方法
"SIGMA" : 0.05, # 高斯展宽参数
}
```
**逻辑分析:**
该代码块设置了VASP计算的参数,包括截断能、k点采样、能量收敛阈值、电子占据态处理方法和高斯展宽参数。这些参数对计算的精度和效率有重要影响。
# 3. DFT在晶体结构分析中的应用
DFT在晶体结构分析中发挥着至关重要的作用,为理解和预测材料的微观结构和性质提供了强大的工具。本章将深入探讨DFT在晶体结构预测、优化、缺陷和表面性质研究中的应用。
### 3.1 晶体结构预测和优化
**3.1.1 晶体结构预测的算法和策略**
晶体结构预测是DFT在材料科学中的一项重要应用。它涉及使用DFT计算来确定给定化学成分的最稳定晶体结构。常用的算法包括:
* **进化算法:**模拟生物进化过程,通过迭代优化来生成候选结构。
* **模拟退火:**从高温开始,逐渐降低温度,允许系统探索不同的结构空间。
* **盆地跳跃:**从一个局部极小值跳跃到另一个,以寻找全局最优结构。
**3.1.2 DFT计算在晶体结构优化中的应用**
一旦预测到候选结构,DFT计算可用于优化其几何形状和原子位置。这涉及使用DFT能量作为目标函数,并使用优化算法(如共轭梯度法或牛顿-拉夫森法)进行迭代优化。通过优化,可以获得具有最低能量和最稳定结构的晶体结构。
### 3.2 晶体缺陷和表面性质的研究
**3.2.1 DFT计算在晶体缺陷研究中的应用**
晶体缺陷是材料中常见的结构不完美性,它们会影响材料的性能。DFT计算可用于研究各种缺陷,包括点缺陷(如空位和间隙原子)、线缺陷(如位错)和面缺陷(如晶界)。通过计算缺陷的形成能、迁移能和电子结构,可以了解缺陷对材料性质的影响。
**3.2.2 DFT计算在表面性质研究中的应用**
材料的表面性质对于其催化、吸附和反应性至关重要。DFT计算可用于研究表面结构、表面能、表面电子态和表面吸附。通过这些计算,可以深入了解材料与环境之间的相互作用,并设计具有特定表面性质的材料。
**代码示例:**
```python
i
```

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
离散傅里叶变换(DFT)是一项强大的数学工具,广泛应用于信号处理、图像处理、语音信号处理、医学成像、气象学、音乐信号处理、电气工程、金融领域、通信工程、计算机视觉、人工智能、生物信息学、材料科学、化学、物理学、机械工程和土木工程等众多领域。
DFT能够将信号从时域分解到频域,揭示信号的频率成分,从而为信号分析和处理提供了宝贵的见解。专栏深入探讨了DFT的原理、提升效率的技巧、在不同领域的应用以及与快速傅里叶变换(FFT)的比较。通过一系列案例研究和实用示例,专栏展示了DFT如何赋能各个行业,从提升信号处理效率到推动科学发现和技术创新。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【线性回归时间序列预测】:掌握步骤与技巧,预测未来不是梦
# 1. 线性回归时间序列预测概述
## 1.1 预测方法简介
线性回归作为统计学中的一种基础而强大的工具,被广泛应用于时间序列预测。它通过分析变量之间的关系来预测未来的数据点。时间序列预测是指利用历史时间点上的数据来预测未来某个时间点上的数据。
## 1.2 时间序列预测的重要性
在金融分析、库存管理、经济预测等领域,时间序列预测的准确性对于制定战略和决策具有重要意义。线性回归方法因其简单性和解释性,成为这一领域中一个不可或缺的工具。
## 1.3 线性回归模型的适用场景
尽管线性回归在处理非线性关系时存在局限,但在许多情况下,线性模型可以提供足够的准确度,并且计算效率高。本章将介绍线
【特征选择工具箱】:R语言中的特征选择库全面解析

# 1. 特征选择在机器学习中的重要性
在机器学习和数据分析的实践中,数据集往往包含大量的特征,而这些特征对于最终模型的性能有着直接的影响。特征选择就是从原始特征中挑选出最有用的特征,以提升模型的预测能力和可解释性,同时减少计算资源的消耗。特征选择不仅能够帮助我
数据清洗的概率分布理解:数据背后的分布特性

# 1. 数据清洗的概述和重要性
数据清洗是数据预处理的一个关键环节,它直接关系到数据分析和挖掘的准确性和有效性。在大数据时代,数据清洗的地位尤为重要,因为数据量巨大且复杂性高,清洗过程的优劣可以显著影响最终结果的质量。
## 1.1 数据清洗的目的
数据清洗
p值在机器学习中的角色:理论与实践的结合

# 1. p值在统计假设检验中的作用
## 1.1 统计假设检验简介
统计假设检验是数据分析中的核心概念之一,旨在通过观察数据来评估关于总体参数的假设是否成立。在假设检验中,p值扮演着决定性的角色。p值是指在原
【品牌化的可视化效果】:Seaborn样式管理的艺术

# 1. Seaborn概述与数据可视化基础
## 1.1 Seaborn的诞生与重要性
Seaborn是一个基于Python的统计绘图库,它提供了一个高级接口来绘制吸引人的和信息丰富的统计图形。与Matplotlib等绘图库相比,Seaborn在很多方面提供了更为简洁的API,尤其是在绘制具有多个变量的图表时,通过引入额外的主题和调色板功能,大大简化了绘图的过程。Seaborn在数据科学领域得
【复杂数据的置信区间工具】:计算与解读的实用技巧
# 1. 置信区间的概念和意义
置信区间是统计学中一个核心概念,它代表着在一定置信水平下,参数可能存在的区间范围。它是估计总体参数的一种方式,通过样本来推断总体,从而允许在统计推断中存在一定的不确定性。理解置信区间的概念和意义,可以帮助我们更好地进行数据解释、预测和决策,从而在科研、市场调研、实验分析等多个领域发挥作用。在本章中,我们将深入探讨置信区间的定义、其在现实世界中的重要性以及如何合理地解释置信区间。我们将逐步揭开这个统计学概念的神秘面纱,为后续章节中具体计算方法和实际应用打下坚实的理论基础。
# 2. 置信区间的计算方法
## 2.1 置信区间的理论基础
### 2.1.1
正态分布与信号处理:噪声模型的正态分布应用解析
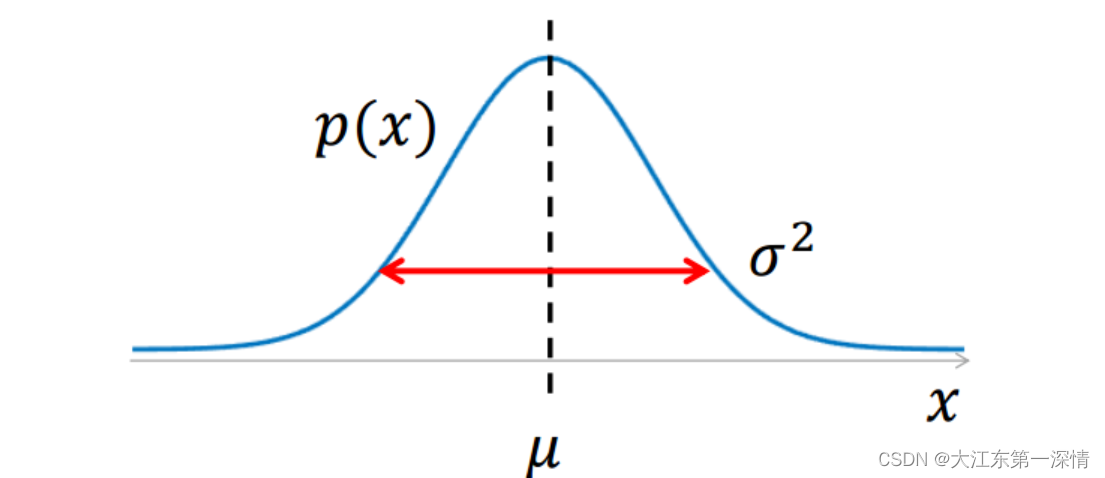
# 1. 正态分布的基础理论
正态分布,又称为高斯分布,是一种在自然界和社会科学中广泛存在的统计分布。其因数学表达形式简洁且具有重要的统计意义而广受关注。本章节我们将从以下几个方面对正态分布的基础理论进行探讨。
## 正态分布的数学定义
正态分布可以用参数均值(μ)和标准差(σ)完全描述,其概率密度函数(PDF)表达式为:
```math
f(x|\mu,\sigma^2) = \frac{1}{\sqrt{2\pi\sigma^2}} e
【时间序列分析】:如何在金融数据中提取关键特征以提升预测准确性
# 1. 时间序列分析基础
在数据分析和金融预测中,时间序列分析是一种关键的工具。时间序列是按时间顺序排列的数据点,可以反映出某
大样本理论在假设检验中的应用:中心极限定理的力量与实践
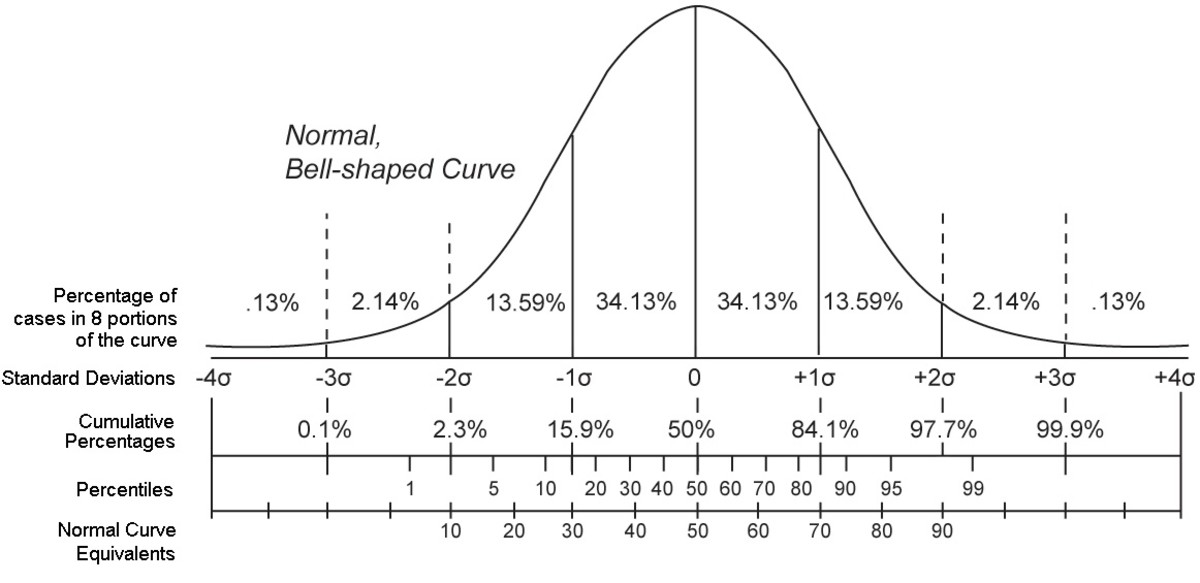
# 1. 中心极限定理的理论基础
## 1.1 概率论的开篇
概率论是数学的一个分支,它研究随机事件及其发生的可能性。中心极限定理是概率论中最重要的定理之一,它描述了在一定条件下,大量独立随机变量之和(或平均值)的分布趋向于正态分布的性
【PCA算法优化】:减少计算复杂度,提升处理速度的关键技术

# 1. PCA算法简介及原理
## 1.1 PCA算法定义
主成分分析(PCA)是一种数学技术,它使用正交变换来将一组可能相关的变量转换成一组线性不相关的变量,这些新变量被称为主成分。
## 1.2 应用场景概述
PCA广泛应用于图像处理、降维、模式识别和数据压缩等领域。它通过减少数据的维度,帮助去除冗余信息,同时尽可能保
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )