Gromacs模拟结果可视化教程:使用VMD和PyMOL展现模拟之美
发布时间: 2024-12-03 08:00:24 阅读量: 16 订阅数: 14 

参考资源链接:[Gromacs模拟教程:从pdb到gro,top文件生成及初步模拟](https://wenku.csdn.net/doc/2d8k99rejq?spm=1055.2635.3001.10343)
# 1. Gromacs模拟结果分析基础
## Gromacs模拟结果分析的重要性
在分子动力学研究领域,Gromacs是一个广泛使用的模拟软件,它能够帮助科学家们理解复杂生物分子系统在原子尺度上的行为。Gromacs模拟的输出文件通常包含了大量数据,包括蛋白质、脂质、核酸等分子的三维结构信息,以及它们随时间变化的轨迹。因此,对这些数据进行分析是必不可少的步骤,以便于我们提取出有科学价值的信息,如蛋白质折叠路径、药物靶点的动态行为或生物大分子之间的相互作用等。
## 基本分析工具介绍
分析Gromacs模拟结果的第一步通常是使用Gromacs自带的工具,如`gmx rms`、`gmx rmsf`和`gmx energy`等,这些工具可以计算均方根偏差(RMSD)、根均方波动(RMSF)和能量项等关键参数。这些参数反映了模拟系统在结构和能量上的特征,是进一步深入分析的基础。
```bash
# 示例:计算蛋白质轨迹的均方根偏差(RMSD)
gmx rms -s topol.tpr -f traj.xtc -o rmsd.xvg
```
在上述命令中,`-s`参数指定了拓扑文件,`-f`参数指定了轨迹文件,`-o`参数指定了输出的RMSD文件。运行此命令后,我们将获得一个包含RMSD值的文件,通常会使用`xmgrace`等工具将其可视化,以便于分析蛋白质结构随时间的变化情况。
## 进阶分析方法概述
虽然Gromacs工具集提供的基础分析功能非常强大,但对于更为深入的可视化和分析,研究者们往往还会借助其他专门的可视化软件,如VMD(Visual Molecular Dynamics)和PyMOL。这些工具可以提供更加直观的三维结构展示,并允许用户在分子层面上进行交互式探索和分析,对于理解模拟结果和撰写高质量的科研论文至关重要。在后续章节中,我们将详细介绍如何使用这些工具来进一步分析Gromacs模拟的结果。
# 2. ```
# 第二章:VMD可视化工具入门
## 2.1 VMD安装与界面介绍
### 2.1.1 安装VMD的系统要求
VMD(Visual Molecular Dynamics)是一个专为分子建模和可视化设计的工具,其系统要求相对比较亲民,可以在多种操作系统上运行,包括Windows、MacOS和Linux。安装VMD之前,需要确认以下几点:
1. **操作系统兼容性**:VMD支持最新的操作系统,以确保最佳的稳定性和性能。用户需要根据其操作系统下载对应版本的安装包。
2. **硬件配置**:虽然VMD对硬件的要求不是特别高,但为了获得流畅的体验,建议至少拥有4GB的RAM以及一个支持图形加速的显卡。
3. **依赖软件**:安装VMD可能需要预先安装一些库文件,如Tcl/Tk,OpenGL等,这些依赖在安装包中通常会自动配置。
### 2.1.2 VMD主界面和菜单结构
VMD的主界面设计简洁,功能区域分布合理。以下是对VMD主界面各部分的简单介绍:
1. **主窗口(Main window)**:这是VMD的中心,用于显示分子结构、轨迹动画等。在这里,用户可以进行各种视觉和交互式操作。
2. **状态栏(Status bar)**:位于窗口底部,显示当前操作的状态信息,如光标坐标、帧数等。
3. **菜单栏(Menu bar)**:包含一系列下拉菜单项,涵盖了VMD几乎所有的功能,如文件操作、显示设置、分子操作等。
VMD菜单栏中的选项主要分为以下几类:
- **文件(File)**:进行文件的打开、保存、导入、导出等操作。
- **显示(Display)**:调整视图设置,控制分子的显示方式。
- **图形(Graphics)**:对分子进行渲染,如调整光线、阴影等。
- **分子(Molecule)**:对加载的分子进行操作,例如载入、删除、编辑等。
- **扩展(Extensions)**:加载额外的分析和可视化插件。
- **窗口(Windows)**:控制VMD中各个子窗口的显示。
- **帮助(Help)**:提供关于VMD的帮助文档和用户指南。
## 2.2 VMD的基本操作与功能
### 2.2.1 导入Gromacs模拟文件
导入Gromacs模拟文件到VMD是一个简单且直接的过程。Gromacs输出的轨迹文件通常有多种格式,包括XTC、TRR、GRO等。以下是导入文件的步骤:
1. 打开VMD。
2. 点击File > New Molecule(文件 > 新建分子)。
3. 在弹出的窗口中,点击Browse(浏览)按钮,找到并选择相应的Gromacs轨迹文件。
4. 根据文件类型,适当配置文件的读取选项,然后点击Load(加载)。
```
mol new example.xtc type xtc
```
该命令会加载名为example.xtc的Gromacs轨迹文件到VMD中。文件类型需要指定为xtc。
### 2.2.2 分子结构的显示与控制
在VMD中,用户可以灵活地控制分子的显示。以下是一些基本的操作:
- **旋转(Rotate)**:使用鼠标左键拖动可以自由旋转视图。
- **缩放(Zoom)**:使用鼠标中键或者鼠标滚轮可以缩放视图。
- **平移(Translate)**:按下Shift键后使用鼠标左键拖动可以平移视图。
在可视化中,控制分子的显示方式也很重要。可以通过Display菜单来更改分子的颜色、渲染方式、光照等属性。
```
graphics top
mol representation CPK
mol color Name
```
以上代码块设置了分子的显示为CPK(查尔斯科德尔-保罗琴)方式,并将颜色设置为按照原子名称显示。
### 2.2.3 颜色编码和标记的设置
VMD允许用户对分子模型进行颜色编码和标记,以突出显示特定的原子或分子。以下是一些操作方法:
- **颜色编码**:通过Color菜单,用户可以选择预设的颜色方案或自定义颜色编码。
- **标记原子或残基**:在Graphics菜单下,使用Labels选项可以为选定的原子或残基添加标记。
```
mol color Name
mol selection "all"
mol representation棍棒
mol addrep 0
```
这段代码块将所有分子的显示样式设置为棍棒模型,并且颜色按照原子名称来编码。
## 2.3 高级可视化技巧
### 2.3.1 动画制作与轨迹分析
VMD不仅可以静态地展示分子结构,还能够制作动画和进行轨迹分析,这在研究分子动力学模拟时非常有用。以下是一些高级操作:
- **动画制作**:用户可以利用VMD的动画控制面板,创建帧序列,调整帧速率,导出动画文件。
- **轨迹分析**:VMD提供了丰富的轨迹分析工具,包括但不限于能量计算、距离测量、角度计算等。
```
animate write movie moviewrite.mpg
```
此命令将制作一个动画文件moviewrite.mpg。
### 2.3.2 分子动力学轨迹的分析与处理
在分子动力学模拟中,轨迹文件记录了整个模拟过程中分子的坐标变化。对这些轨迹文件的分析可以帮助我们理解分子运动和变化的
```

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏目录
最低0.47元/天 解锁专栏
买1年送1年
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
状态机与控制单元:Logisim实验复杂数据操作管理
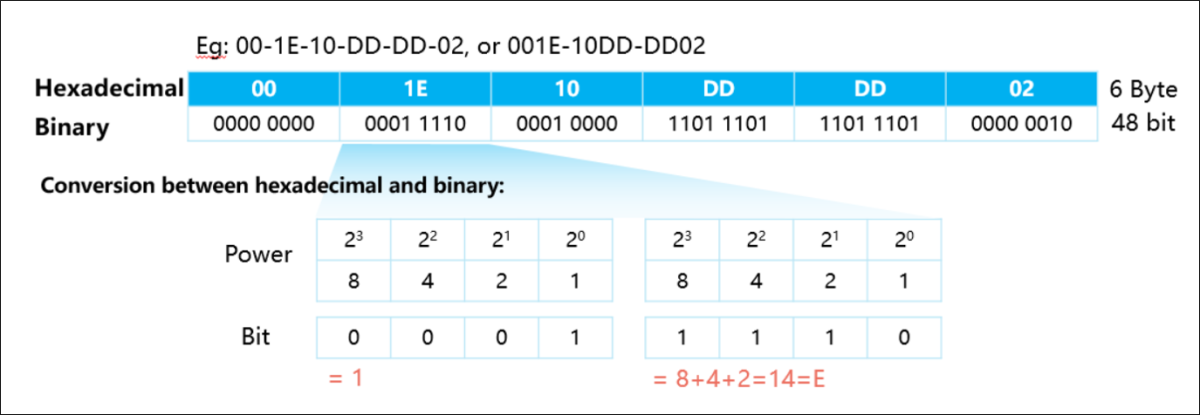
参考资源链接:[Logisim实验教程:海明编码与解码技术解析](https://wenku.csdn.net/doc/58sgw98wd0?spm=1055.2635.3001.10343)
# 1. 状态机与控制单元的理论基础
状态机是一种计算模型,它能够通过一系列状态和在这些状态之间的转移来表示对象的行为。它是控制单元设计的核心理论之一,用于处理各种
MT7981硬件加速功能:4种方法发挥硬件最大潜力
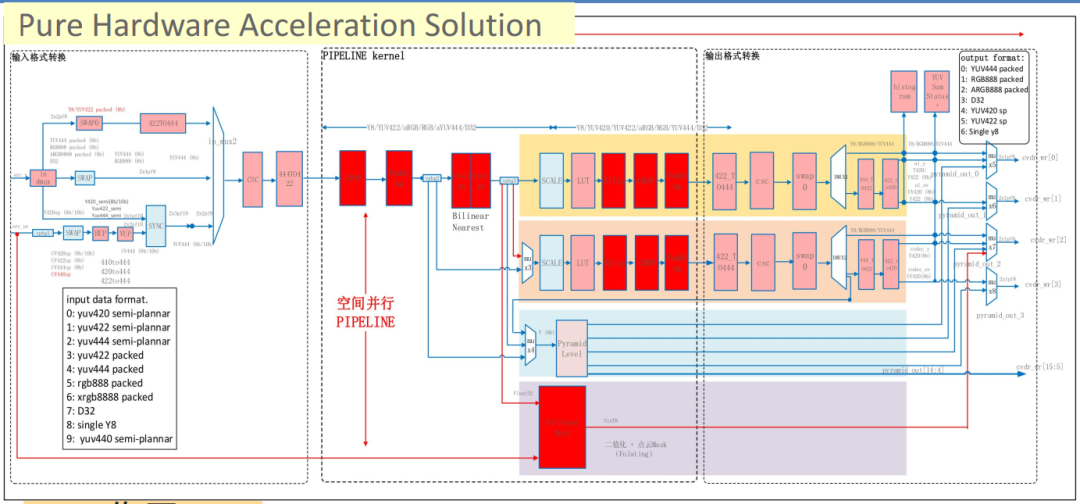
参考资源链接:[MT7981数据手册:专为WiFi AP路由器设计的最新规格](https://wenku.csdn.net/doc/7k8yyvk5et?spm=1055.2635.3001.10343)
# 1. MT7981硬件加速功能概述
随着现代技术的快速发展,硬件加速成为了提升性能的关键因素之一。MT7981作为
数字电路设计自动化与智能化:未来趋势与实践路径

参考资源链接:[John F.Wakerly《数字设计原理与实践》第四版课后答案汇总](https://wenku.csdn.net/doc/7bj643bmz0?spm=1055.2635.3001.10343)
# 1. 数字电路设计自动化与智能化概述
数字电路设计自动化与智能化是现代电子设计领域的两大重要趋势,它们极大地提升了设计效
软件工程中的性能优化:理论结合案例,助你打造极速软件

参考资源链接:[吕云翔《软件工程-理论与实践》习题答案解析](https://wenku.csdn.net/doc/814p2mg9qb?spm=1055.2635.3001.10343)
# 1. 性能优化的基本概念
在信息技术迅猛发展的今天,性能优化已成为确保系统高效运行的关键组成部分。在这一章中,我们将探讨性能优化的
【HOLLiAS MACS V6.5.2数据采集与分析】:实时数据驱动决策的力量
参考资源链接:[HOLLiAS MACS V6.5.2用户操作手册:2013版权,全面指南](https://wenku.csdn.net/doc/6412b6bfbe7fbd1778d47d3b?spm=1055.2635.3001.10343)
# 1. HOLLiAS MACS V6.5.2概述及
CPCI标准术语集:破解专业词汇,提升应用精准度

参考资源链接:[CPCI标准规范中文版.pdf](https://wenku.csdn.net/doc/645f33b65928463033a7b79b?spm=1055.2635.3001.10343)
# 1. CPCI标准术语集概述
CPCI标准术语集是一套旨在统一计算机、信息技术及相关学科中使用的专业术语的规范。本章将对CPCI
【光刻技术的未来】:从传统到EUV的技术演进与应用
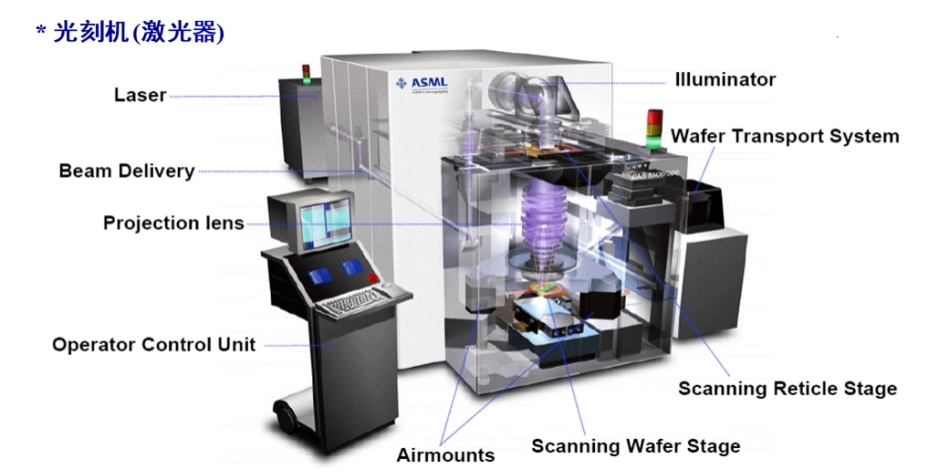
参考资源链接:[Fundamentals of Microelectronics [Behzad Razavi]习题解答](https://wenku.csdn.net/doc/6412b499be7fbd1778d40270?spm=1055.2635.3001.10343)
# 1. 光刻技术概述
## 1.1 光刻技术简介
光刻技术是半导体制造中不可或缺的工艺,它使用光学或电子束来在硅片表面精确地复
【PitStop Pro 2019效率革命】:掌握高级功能,立竿见影提升PDF编辑

参考资源链接:[Enfocus PitStop Pro 2019:全面指南与强大功能详解](https://wenku.csdn.net/doc/6412b6bebe7fbd1778d47d28?spm=1055.2635.3001.10343)
# 1. PitStop Pro 2019概览
PitStop Pro 2019是Enf
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送1年
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )