GROMACS模拟流程全解析:从零开始到成功运行的必备步骤
发布时间: 2024-12-01 09:35:36 阅读量: 7 订阅数: 12 

参考资源链接:[Gromacs模拟教程:从pdb到gro,top文件生成及初步模拟](https://wenku.csdn.net/doc/2d8k99rejq?spm=1055.2635.3001.10343)
# 1. GROMACS模拟概述
## 1.1 GROMACS简介
GROMACS(GROningen MAchine for Chemical Simulations)是一款著名的开源分子动力学模拟软件,广泛应用于生物分子系统的模拟,如蛋白质、脂质膜和核酸等。它的高效性能使其成为计算化学和生物物理学领域不可或缺的工具之一。
## 1.2 分子动力学模拟基础
分子动力学模拟是一种利用经典力学方程来模拟粒子(如原子和分子)运动的方法。该技术能够预测分子系统的宏观性质,通过模拟的时间演化来理解微观层面的物理和化学过程。
## 1.3 GROMACS在研究中的应用
GROMACS被大量应用于药物设计、生物材料研究以及蛋白质折叠等领域。它支持多种力场,能够进行系统的能量最小化、平衡以及长时间的轨迹生成,是科研人员探索生物分子复杂行为的重要工具。
# 2. GROMACS安装和配置
### 2.1 系统要求和安装步骤
GROMACS是一个功能强大的分子动力学模拟软件,广泛应用于生物物理、化学和材料科学等领域。为了确保软件的稳定运行,首先需要了解其对系统的基本要求,并按照合适的步骤进行安装。
#### 2.1.1 硬件和软件的准备
在安装GROMACS之前,需要准备好一台满足最低系统要求的计算机。GROMACS对硬件的要求主要包括处理器、内存、磁盘空间和操作系统。以GROMACS 2021版本为例,对于处理器至少需要支持SSE4.1指令集;内存和磁盘空间应根据模拟系统复杂度进行配置,一般至少需要4GB内存和10GB磁盘空间。操作系统方面,GROMACS支持多数版本的Linux和Windows操作系统,以及MacOS。
对于软件环境,需要预先安装CMake、编译器(如gcc/g++),以及数学库如MKL或OpenBLAS。建议操作系统使用基于Debian或Red Hat的发行版,如Ubuntu或CentOS,因为GROMACS官方网站提供了针对这些系统的预编译二进制文件。
#### 2.1.2 安装GROMACS的环境配置
安装GROMACS时,可以通过预编译的二进制文件安装,也可以从源代码编译安装。如果选择编译安装,可以使用以下命令来配置环境并进行编译:
```bash
# 下载GROMACS源代码包
wget https://ftp.gromacs.org/pub/gromacs/gromacs-2021.tar.gz
# 解压源代码包
tar xfz gromacs-2021.tar.gz
# 进入解压后的目录
cd gromacs-2021
# 配置安装选项
cmake -DGMX_BUILD_OWN_FFTW=ON -DREGRESSIONTEST_DOWNLOAD=ON -DCMAKE_INSTALL_PREFIX=/usr/local/gromacs-2021 .
# 编译安装
make
make install
```
配置选项中`-DGMX_BUILD_OWN_FFTW=ON`指定了编译内置的FFT库,`-DREGRESSIONTEST_DOWNLOAD=ON`用于下载回归测试数据,`-DCMAKE_INSTALL_PREFIX`用于指定安装路径。
### 2.2 GROMACS版本的选择与升级
#### 2.2.1 不同版本的对比和应用场景
GROMACS每年都会更新若干版本,每个版本都会增加新的功能和改进。对于研究人员而言,选择合适的版本对于模拟工作的成功至关重要。较新版本通常修复了旧版本的bug,提供了更高效的计算速度和更强大的功能。例如,GROMACS 2020版本改进了TNG格式的支持,增强了并行处理能力,而GROMACS 2021版本进一步优化了GPU加速性能。
不同版本的GROMACS也可能会有API或数据格式的变更,因此需要根据团队的工作流和已有的配置文件选择合适的版本。如果已经习惯了使用某个版本的GROMACS,除非必要,否则一般不需要频繁升级。
#### 2.2.2 升级流程和注意事项
如果决定升级GROMACS,以下是一些基本的升级流程和注意事项:
1. **备份旧版本数据**:在升级前,备份任何重要的配置文件、模拟数据和运行日志。
2. **卸载旧版本**:根据安装方式,找到并卸载当前版本的GROMACS。如果是通过包管理器安装,使用相应的卸载命令;如果是从源代码编译安装,删除安装目录即可。
3. **安装新版本**:按照上述步骤安装新的GROMACS版本。
4. **验证安装**:使用简单的模拟测试确认新版本的GROMACS是否正常工作。
5. **数据迁移**:如果有必要,根据新版本的API或数据格式变更,调整原有的脚本或输入文件。
6. **新旧版本并存**:如果不确定新版本的稳定性,可以选择新旧版本同时安装,并在确认新版本工作正常后逐步切换。
### 2.3 GROMACS并行计算设置
#### 2.3.1 多核CPU与GPU加速配置
为了提高模拟效率,GROMACS支持多核CPU并行计算以及GPU加速。正确配置并行计算环境,可以让模拟工作更高效。
对于CPU并行,主要通过设置环境变量`GMX_OPENMP`来指定使用的线程数。例如,要在4核CPU上运行模拟,可以在运行脚本前设置:
```bash
export GMX_OPENMP=4
```
对于GPU加速,需要确保系统已经安装了支持CUDA或OpenCL的显卡驱动和运行库。在GROMACS的配置过程中,应启用GPU支持选项:
```bash
cmake -DGMX_GPU=CUDA -DGMX_BUILD_OWN_FFTW=ON ..
```
#### 2.3.2 跨节点计算环境搭建
在进行大规模模拟时,可能需要在多个计算节点之间分配任务。GROMACS支持通过MPI(Message Passing Interface)进行跨节点的并行计算。搭建跨节点计算环境通常需要一个支持MPI的高性能计算集群。
搭建过程中,需要在每个节点上安装MPI库并配置好网络通信。安装完成后,通过MPI运行程序来启动跨节点并行计算:
```bash
mpirun -np <总的核心数> gmx mdrun -s <拓扑文件> -o <输出文件> -c <坐标文件> -g <日志文件>
```
其中`-np`参数后跟的是所有节点上可用的核心数之和。
以上就是关于GROMACS安装和配置的基本介绍,为确保模拟的成功进行,系统准备、版本选择、并行计算设置都是必须要细心处理的环节。接下来,我们将深入了解模拟前的准备工作。
# 3. GROMACS模拟前的准备工作
## 3.1 模拟系统的构建
### 3.1.1 选择合适的力场
在开始构建模拟系统之前,首先需要选择一个适合您研究体系的力场(force field)。力场决定了原子间的相互作用和参数,不同的力场在参数化时考虑了不同的物理特性,因此对模拟结果有显著的影响。GROMACS支持多种力场,如AMBER、CHARMM、OPLS-AA等。
选择力场时应考虑以下几个因素:
- **研究体系**:力场通常是为特定类型的分子系统设计的,例如蛋白质、核酸、有机小分子或金属离子。
- **所需精度**:一些力场如AMBER提供了不同的版本,以适应不同研究需求的精度和计算效率。
- **兼容性**:所选力场需要与您的模拟类型兼容,如温度控制、压力模拟、离子条件等。
在GROMACS中,力场参数文件通常以`.itp`或`.ff`为后缀名,可以在GROMACS安装目录中的`share/gromacs/top`文件夹中找到。
### 3.1.2 分子结构的获取和处理
一旦确定了力场,下一步是获取和处理您的研究体系分子结构。您可能需要从蛋白质数据库(如PDB)下载结构文件,并使用相关软件如Pymol或VMD进行预处理。
### 操作步骤
1. 下载PDB文件。
2. 使用Pymol去除水分子、金属离子等非目标分子。
3. 为缺失的原子、残基添加缺失的重原子。
4. 对残基进行质子化状态的校正,以匹配生理pH值。
5. 保存处理后的结构文件,通常为`.gro`或`.pdb`格式。
## 3.2 模拟盒子的设置和溶剂化
### 3.2.1 确定模拟盒子的大小和形状
模拟盒子定义了模拟系统的空间边界。在构建时需要考虑所研究分子的大小、形状以及预期的模拟条件。例如,对于蛋白质模拟,通常使用正方体或长方体盒子来包含整个分子并确保在模拟过程中有足够的空间进行运动。
### 操作步骤
1. 使用`editconf`命令来创建模拟盒子,调整大小和形状。
```bash
editconf -f protein.gro -o box.gro -bt cubic -d 1.0
```
这里`-f`参数指定输入文件,`-o`参数指定输出文件,`-bt`指定盒子类型为正方体,`-d`指定盒子边缘到分子的最小距离。
2. 在上述命令中,`-d`参数设置为1.0纳米,这意味着蛋白质周围至少有1纳米的空间。
### 3.2.2 溶剂分子的添加和最小化
在定义好模拟盒子后,下一步是添加溶剂分子。常用的溶剂模型有TIP3P、TIP4P等。溶剂分子填充盒子后,通常需要进行能量最小化步骤以释放分子间的过紧接触。
### 操作步骤
1. 使用`genbox`命令添加溶剂分子。
```bash
genbox -cp box.gro -cs spc216.gro -o solvated_box.gro -p topol.top
```
其中`-cp`指定蛋白质盒子文件,`-cs`指定溶剂的配置文件,`-o`指定输出文件,`-p`指定拓扑文件。
2. 使用`grompp`准备能量最小化输入文件。
```bash
grompp -f minim.mdp -c solvated_box.gro -p topol.top -o em.tpr
```
这里`-f`指定最小化参数文件,`-c`指定输入的结构文件。
3. 使用`mdrun`运行能量最小化。
```bash
mdrun -v -deffnm em
```
`-v`表示详细模式,`-deffnm`指定输出文件名的前缀。
## 3.3 能量最小化和平衡步骤
### 3.3.1 能量最小化的必要性
在GROMACS模拟之前,能量最小化(Energy Minimization)是一个关键步骤。其目的是减少初始结构中的高能量状态,避免在分子动力学模拟的早期阶段出现原子剧烈移动,影响模拟的收敛性。
### 操作步骤和分析
1. **准备最小化参数文件**(minim.mdp),通常包含以下几个关键参数:
- `emtol`:能量梯度的收敛标准,默认值为1000千焦耳/摩尔。
- `nsteps`:最小化步数,通常足够以达到收敛。
2. **运行最小化**:
- 命令行中使用`grompp`生成可运行的输入文件(.tpr)。
- 使用`mdrun`执行实际的能量最小化。
3. **结果分析**:
- 查看能量最小化的输出日志文件,确认是否达到收敛标准。
- 使用`gmx energy`查看不同能量项随步骤的变化情况。
### 3.3.2 系统热平衡的策略和方法
在能量最小化之后,系统需要进行热平衡(NVT和NPT平衡)以确保温度和压力达到预定条件。这个过程称为平衡步骤,目的是让系统达到实际物理条件下的平衡状态。
### 操作步骤和分析
1. **准备NVT平衡参数文件**(nvt.mdp),这里需要设置:
- `gen-vel`:生成初始速度。
- `ref-t`:目标温度。
2. **准备NPT平衡参数文件**(npt.mdp),需要添加:
- `ref-p`:目标压力。
- `compressibility`和`isothermal-compressibility`:系统压缩率和等温压缩率。
3. **执行平衡步骤**:
- 使用`grompp`和`mdrun`依次运行NVT和NPT平衡。
- 通过输出日志文件检查系统是否达到平衡状态。
4. **结果分析**:
- 使用`gmx energy`监控温度、压力、能量等物理量是否达到目标值。
- 使用`gmx rmsd`和`gmx rmsf`检查系统结构是否稳定。
# 4. GROMACS模拟执行和分析
## 4.1 运行模拟和参数设置
### 4.1.1 长时间动力学模拟的参数配置
长时间动力学模拟是分子动力学研究中的重要环节,它能够提供系统随时间演化的详细信息。为了确保模拟的稳定性和物理准确性,需要进行细致的参数配置。
在GROMACS中,模拟参数主要通过拓扑文件(.top)和模拟参数文件(.mdp)来设置。对于长时间动力学模拟,特别需要注意以下参数:
- **时间步长(dt)**:这是模拟中每一个步骤所代表的实际时间长度。对于长时间模拟来说,推荐使用较短的时间步长以保证数值稳定性,通常为2飞秒(fs)。
- **积分器**:选择合适的积分器对于长时间稳定模拟至关重要。常用的积分器有leap-frog和Velocity Verlet,后者在处理约束系统时更为稳定。
- **总模拟时间(nsteps)**:这是模拟将要进行的总步数,对应实际时间可以通过步长来计算。对于长时间模拟,需要根据系统稳定性和研究需求谨慎选择。
- **保存频率**:决定输出结果(如位置、速度、能量等)的频率,频繁保存会增加磁盘I/O消耗,但对于分析和监控至关重要。
- **温度和压力耦合**:长时间模拟往往需要系统达到热平衡和压力平衡。温度耦合(tcoupl)和压力耦合(pcoupl)参数的选择,以及耦合时间常数(tau-t/tau-p)的设定,都会影响系统达到平衡的速度和稳定性。
### 4.1.2 温度、压力控制的原理和应用
温度和压力控制是确保模拟过程稳定的关键。GROMACS提供多种温度和压力耦合算法来模拟不同环境下的物理系统。
#### 温度控制:
- **恒温器(Thermostat)**:恒温器的目的是维持模拟系统的温度在一个预设值,常用的恒温器有Berendsen、Velocity rescale和Nosé-Hoover等。
- **Berendsen恒温器**:使用指数衰减的方式调节系统温度,适用于平衡阶段的模拟,但在产生正确热力学量方面有局限性。
- **Velocity rescale恒温器**:通过调节分子速度来控制温度,它是一个改进的Berendsen恒温器,保持了较好的热力学性质,适合用于平衡和生产阶段。
- **Nosé-Hoover恒温器**:它能产生正确的热力学分布,适用于需要精确控制温度和模拟长时间动力学的场景。
在参数文件中,温度耦合组(tc-grps)需要指定哪些粒子组需要进行温度控制。温度耦合时间常数(tau-t)决定了温度调整的速度,它需要根据系统的具体特性来设置。
```groovy
; Example temperature coupling parameters in the .mdp file
tcoupl = Nose-Hoover
tc-grps = Protein Non-Protein
tau_t = 0.5 0.5
ref_t = 300 300
```
#### 压力控制:
- **压强耦合**:类似于温度控制,压强耦合用于控制模拟体系的体积,从而控制其压强。常用的压强耦合算法有Berendsen、Parrinello-Rahman等。
- **Berendsen压强耦合**:它适用于快速达到系统平衡的情况,但不适合长时间模拟,因为它会偏离实际的等温等压状态。
- **Parrinello-Rahman压强耦合**:这是一种更加物理的方法,适合长时间的生产运行,因为它能够在保持正确热力学性质的同时进行压强控制。
在选择压强耦合算法时,耦合组(pc-grps)、耦合时间常数(tau-p)、目标压强(ref-p)等参数需要被相应地配置。
```groovy
; Example pressure coupling parameters in the .mdp file
pcoupl = Parrinello-Rahman
pcoupltype = isotropic
tau_p = 5.0
compressibility = 4.5e-5
ref_p = 1.0
```
在实际应用中,选择合适的温度和压力耦合算法以及参数配置,需要根据研究目的和系统特性来综合考虑。
## 4.2 模拟数据的监控和日志分析
### 4.2.1 关键指标的实时监控方法
长时间的分子动力学模拟会产生大量的数据,对关键指标的实时监控是确保模拟有效性的关键步骤。GROMACS通过日志文件(如 log.md,能量文件 ener.edr)来记录模拟过程中的关键参数。
实时监控通常会关注以下指标:
- **温度**:监测系统是否达到预期的温度值。
- **压力**:检查系统是否达到或稳定在设定的压强。
- **能量**:分析系统的总能量、动能、势能等,确保能量守恒。
- **密度**:对于溶剂化体系,检查模拟盒子的密度是否稳定。
- **配位数**:对于固体材料或大分子系统,监测原子间的配位情况。
在GROMACS中,可以使用`gmx monitor`命令实时监控这些指标。该命令提供了一个交互式界面来展示关键物理量,如下所示:
```shell
gmx monitor
```
执行该命令后,将显示一个实时更新的窗口,其中包括了温度、压力、能量、密度等指标。用户可以自定义显示的内容,从而专注于特定的模拟参数。
### 4.2.2 日志文件的解读和故障排除
模拟完成后,GROMACS生成的日志文件包含了丰富的信息,这些信息对于分析模拟过程、理解可能出现的问题至关重要。
- **能量文件(ener.edr)**:该文件包含了模拟过程中能量项的详细记录。可以使用`gmx能量`命令来分析能量随时间的变化:
```shell
gmx energy -f ener.edr
```
这将打开一个交互式界面,允许用户选择感兴趣的能量项,并输出相应的图像或数据。
- **日志文件(log.md)**:日志文件记录了模拟运行的详细信息,包括警告和错误。对日志文件的分析可以从以下几个方面入手:
- **稳定性分析**:检查能量是否随时间稳定,如果能量大幅波动,则可能存在稳定性问题。
- **错误检查**:搜索“WARNING”和“ERROR”关键字,快速定位可能的问题,如不合理的力场参数或约束失败。
- **性能评估**:查看模拟的时间步长,确定是否达到了设定的数值稳定性条件。
- **检查点文件(.cpt)**:在长时间模拟中,GROMACS会定期保存检查点文件,以便在意外中断后能够从最近的检查点恢复模拟。检查检查点文件是否按预期生成,可以帮助确定模拟是否顺利进行。
通过上述方法,可以系统地分析GROMACS模拟的输出文件,从而对模拟的有效性进行评估,并对潜在的问题进行诊断。
## 4.3 结果分析和可视化工具
### 4.3.1 GROMACS自带分析工具的使用
GROMACS自带了多种分析工具,可以直接在命令行中使用,这些工具涵盖了从基本的统计分析到复杂的生物物理量计算。
#### 基本统计分析
GROMACS中的`gmx能量`和`gmx rms`是最基础的分析工具,可以用来计算能量、配位数和根均方偏差(RMSD)等统计参数。
- `gmx energy`:分析能量文件,输出能量随时间的变化。
- `gmx rms`:计算结构的均方根偏差(RMSD),并比较不同结构之间的相似性。
#### 高级分析
GROMACS提供了专门的分析工具来处理更复杂的数据:
- `gmx gyrate`:计算模拟盒子中分子的半径,用于分析蛋白质结构的紧凑性。
- `gmx sasa`:分析溶剂可及表面积(SASA),来了解蛋白质表面暴露程度。
- `gmx hbond`:检测和分析氢键的形成和断裂。
例如,以下是一个计算和绘制氢键统计的脚本:
```shell
gmx hbond -f traj.xtc -s topol.tpr -num hbnum.xvg
```
该命令将输出文件`hbnum.xvg`,它包含了随时间变化的氢键数量信息。
### 4.3.2 第三方可视化软件的配合使用
除了GROMACS自带的工具外,第三方可视化软件为模拟结果提供了更加丰富的可视化选项。
- **VMD(Visual Molecular Dynamics)**:一个强大的分子可视化工具,可以导入GROMACS输出的轨迹文件(如`.xtc`或`.gro`),进行详细的结构和动力学分析,并支持渲染和动画制作。
- **PyMOL**:一个广泛使用的分子图形系统,特别适用于蛋白质结构的展示。PyMOL可以直接读取GROMACS的结构文件,支持多种显示模式和复杂的结构比较。
- **RasMol**:一个简单的分子可视化工具,虽然功能不如VMD和PyMOL强大,但其易用性和速度对于快速检查蛋白质结构非常有帮助。
例如,将GROMACS的轨迹文件导入VMD,并进行基本的可视化操作的步骤如下:
```tcl
mol new topol.gro
animate read xtc traj.xtc
```
这将创建一个新分子,并读取轨迹文件进行动画展示。
为了进行更高级的可视化分析,如绘制蛋白质的RMSD分布图,可以将`gmx rms`的输出导入到R或Python脚本中,并使用绘图库(如matplotlib)进行详细的数据展示:
```python
import matplotlib.pyplot as plt
# 读取 RMSD 数据
rmsd_data = np.loadtxt('rmsd.xvg', comments=['#'])
# 绘制 RMSD 图
plt.plot(rmsd_data[:,0], rmsd_data[:,1])
plt.xlabel('Time (ps)')
plt.ylabel('RMSD (nm)')
plt.title('RMSD of Protein Structure')
plt.show()
```
以上可视化工具和脚本共同构成了GROMACS模拟结果分析的强大生态系统,它们相辅相成,为研究者提供从基本数据处理到深入科学解释的完整工具链。
# 5. GROMACS高级应用和案例分析
在前四章中,我们对GROMACS的基本概念、安装配置、准备工作以及如何执行模拟和分析结果有了深入的理解。本章将深入探讨GROMACS在高级应用和案例分析方面的内容。我们将探讨复杂系统的模拟策略、优化技巧以及通过实际案例展示模拟流程,以帮助读者更好地将GROMACS应用到实际研究中。
## 5.1 复杂系统的模拟策略
在模拟复杂的生物物理系统时,如蛋白质-配体相互作用或大分子聚合物,需要特别关注模拟的策略,以确保结果的准确性和效率。
### 5.1.1 蛋白质-配体相互作用模拟
模拟蛋白质与小分子配体的相互作用是药物设计领域的常见任务。为了精确模拟这种相互作用,需要对力场参数进行仔细的选择,特别是对于配体分子。如果配体分子不在标准力场参数库中,可能需要进行量子力学计算,以获得适当的力场参数。
**操作步骤示例:**
1. 使用量子化学软件计算配体分子的几何结构和电荷分布。
2. 根据计算结果,手动设置或使用力场参数生成工具来调整配体的力场参数。
3. 在GROMACS中构建蛋白质和配体复合物模型。
4. 运行能量最小化和平衡步骤,确保系统稳定。
5. 执行长时间的MD模拟,并监控关键的蛋白质-配体相互作用。
### 5.1.2 大分子聚合物的模拟要点
大分子聚合物的模拟通常需要大量的计算资源。为了使模拟更加高效,可以采取一些策略:
1. **聚合度的选择:** 需要平衡聚合物链的长度与计算成本之间的关系,选取合适聚合度的模型。
2. **非键相互作用的截断:** 合理设置范德瓦尔斯和库仑相互作用的截断距离,以减少计算负担。
3. **多重时间步长:** 在可能的情况下,使用多重时间步长积分算法可以节省计算时间。
4. **并行计算:** 充分利用高性能计算资源,进行有效的并行计算,加速模拟进程。
**代码块示例:**
```gromacs
; grompp.mdp - 运行GROMACS预处理的输入文件示例
title = Polymer Simulation
cpp = /lib/cpp
include = -I../top
define = -DPOSRES
; 非键相互作用的设置
cutoff-scheme = Verlet
rvdw = 1.2
rcoulomb = 1.2
; 时间步长和输出设置
dt = 0.002
nsteps = 50000
; 使用多重时间步长算法
nstcomm = 100
```
## 5.2 GROMACS模拟的优化技巧
随着模拟系统的复杂性增加,优化模拟以提升性能和速度变得愈发重要。以下是一些优化模拟的技巧:
### 5.2.1 性能提升的策略
1. **硬件优化:** 使用高性能的CPU和GPU进行计算。
2. **软件优化:** 确保GROMACS的版本是最新的,并且根据需要选择合适的编译选项。
3. **并行计算优化:** 根据计算资源,合理分配任务到不同的CPU核心或GPU。
4. **算法选择:** 根据系统特性选择合适的积分算法和并行算法,如PME(粒子网格Ewald)电荷计算。
### 5.2.2 模拟精度和速度的权衡
为了保证模拟的精度,通常需要较小的时间步长和较长的模拟时间,这会增加计算成本。因此,必须在模拟精度和计算速度之间找到平衡点。
**参数示例:**
- 对于大多数蛋白质系统,时间步长可以设为2 fs,总模拟时间可能在100 ns以上。
- 对于较大的系统,如膜蛋白,时间步长可能需要减小至1 fs,模拟时间也相应增加。
## 5.3 实际案例的模拟流程展示
在本小节中,我们将通过一个具体项目来展示模拟流程,同时评估模拟结果,并给出反馈。
### 5.3.1 一个具体项目的模拟流程案例
假设我们正在进行一个关于病毒衣壳蛋白的项目,我们的目标是了解其结构的稳定性。以下是模拟流程的简要概述:
1. **建模:** 获取病毒衣壳蛋白的三维结构,并导入GROMACS。
2. **能量最小化:** 进行能量最小化以消除任何潜在的构象应力。
3. **平衡:** 进行NVT(等温等容)和NPT(等温等压)平衡,以达到热力学和力学的稳定状态。
4. **生产运行:** 在平衡的基础上进行长时间的MD模拟,收集数据。
### 5.3.2 效果评估与总结反馈
模拟完成后,需要对模拟过程进行评估,判断模拟是否达到预期的效果。这可能包括:
- **结构稳定性分析:** 通过比较模拟前后的结构来评估蛋白的稳定性。
- **动力学行为分析:** 使用RMSD(均方根偏差)和RMSF(均方根波动)分析蛋白质残基的运动。
- **相互作用分析:** 分析蛋白质内部或与溶剂之间的相互作用。
通过这些分析,我们可以得到模拟的反馈,并据此调整模拟参数或策略,以便进行下一轮的模拟工作。这样的循环优化过程对于提升模拟质量和理解复杂的生物物理过程至关重要。

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
GROMACS模拟流程专栏深入探讨了GROMACS模拟的各个方面,从基础概念到高级技术。专栏包含一系列文章,涵盖以下主题:
* 提升模拟速度的硬件和参数优化技巧
* 处理复杂系统的进阶指南
* 性能瓶颈和高效优化策略
* 轨迹文件解读和分析秘籍
* 并行计算的应用技巧
* 能量最小化原理和步骤
* 温度和压力控制原理和技巧
* 溶剂模型选择和优化技巧
* 质心限制技术的应用和释放
* 周期性边界条件的解析
* 反应场方法的理论和应用
* 非键相互作用的截断和长程修正技巧
* 约束算法的选择和优化
* 恒定体积和恒定压力的比较和选择
* 自由能计算的方法和技巧
该专栏旨在为GROMACS用户提供全面的指南,帮助他们优化模拟流程,解决复杂问题,并获得准确可靠的结果。
专栏目录
最低0.47元/天 解锁专栏
买1年送1年
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
ESO在故障检测与诊断中的作用:策略与方法

参考资源链接:[自抗扰控制技术解析:扩张状态观测器(ESO)与参数整定](https://wenku.csdn.net/doc/1uuy08s1i3?spm=1055.2635.3001.10343)
# 1. ESO在故障检测与诊断中的重要性
在现代IT系统和工业自动化中,故障检测与诊断是确保系统稳定运行和延长设备寿命的关键环节。ESO(Extended State O
MapMatrix3D性能优化:大数据量下保持性能的秘密武器

参考资源链接:[航天远景MapMatrix3D测图操作记录.doc](https://wenku.csdn.net/doc/6412b786be7fbd1778d4a9b1?spm=1055.2635.3001.10343)
# 1. MapMatrix3D简介与性能挑战
MapMatrix3D是一款广泛应用
【MATLAB结构优化】:数据类型与结构的高级管理
参考资源链接:[Simulink学习笔记:断路器控制与信号流连接解析](https://wenku.csdn.net/doc/6s79esxwjx?spm=1055.2635.3001.10343)
# 1. MATLAB结构优化概述
MATLAB作为一款高性能的数学软件,广泛应用于工程计算、算法开发、数据可视化等领域。结构优化作为其核心功能之一,涉及到算法、数据结构以及程序设计等多个层面的高效实现。本章节旨在为读者提供MATLAB结构优化的入门指导,让读者能够快速了解和掌握MATLAB在结构优化方面的基本概念、原理和应用方法。
我们将从结构优化的基本概念入手,简要概述其在工程设计与软件开
INA226与无线传感网络集成:物联网(IoT)时代的智能连接

参考资源链接:[INA226:I2C接口电流电压功率监控器详解](https://wenku.csdn.net/doc/644b80f9ea0840391e559828?spm=1055.2635.3001.10343)
# 1. INA226与无线传感网络
嵌入式系统集成VITA 42.0 XMC模块:一步到位的解决方案
参考资源链接:[ANSI/VITA 42.0-2008(R2014) XMC标准规范详解](https://wenku.csdn.net/doc/6401ad34cce7214c316eeac0?spm=1055.2635.3001.10343)
# 1. VITA 42.0 XMC模块概述
XMC(Express Mezzanine Card)模块作为基于VITA 42.0标准的扩展卡,为嵌入式计算机系统提供了灵活而强大的扩展能力。这种模块通过提供高速串行接口和多功能I/O,使得系统在保持紧凑尺寸的同时,仍能满足高性能计算的需求。
## 1.1 XMC模块的市场定位和应用价值
XMC模块
Innovus文本命令创新:跨领域应用案例深度解析

参考资源链接:[Innovus 21.13文本命令参考:完整指南](https://wenku.csdn.net/doc/35a5bnk8vy?spm=1055.2635.3001.10343)
# 1. Innovus文本命令的基础与原理
## 1.1 Innovus文本命令简介
Innovus是Cadence公司推出的一款先进的IC物理设计工具,其操
热循环测试速成:JEDEC JESD47L:2022产品测试教程

参考资源链接:[2022年JEDEC JESD47L:集成电路应力测试驱动的验收标准详解](https://wenku.csdn.net/doc/1meq3b9wrb?spm=1055.2635.3001.10343)
# 1. 热循环测试概述与标准解读
## 1.1 热循环测试的基本概念
热循环测试是一种评估材料、设备或系统在重复经历热应力作用下的性能和可靠性的方法。通
【存储解决方案】:AFBC在SSD_HDD中的性能对比与应用案例

参考资源链接:[AFBC:ARM帧缓冲压缩技术详解](https://wenku.csdn.net/doc/5h2zjv85x7?spm=1055.2635.3001.10343)
# 1. 存储技术的基础概念
## 1.1 数据存储的基本原理
存储技术是信息技术的核心组成部分之一,其主要功能是持久保存数据,为计算设备提供数据读写服务。数据存储的基础原理涉及到数据的编码、存
【设计迭代新策略】:LS-PrePost优化设计方法的全面解析
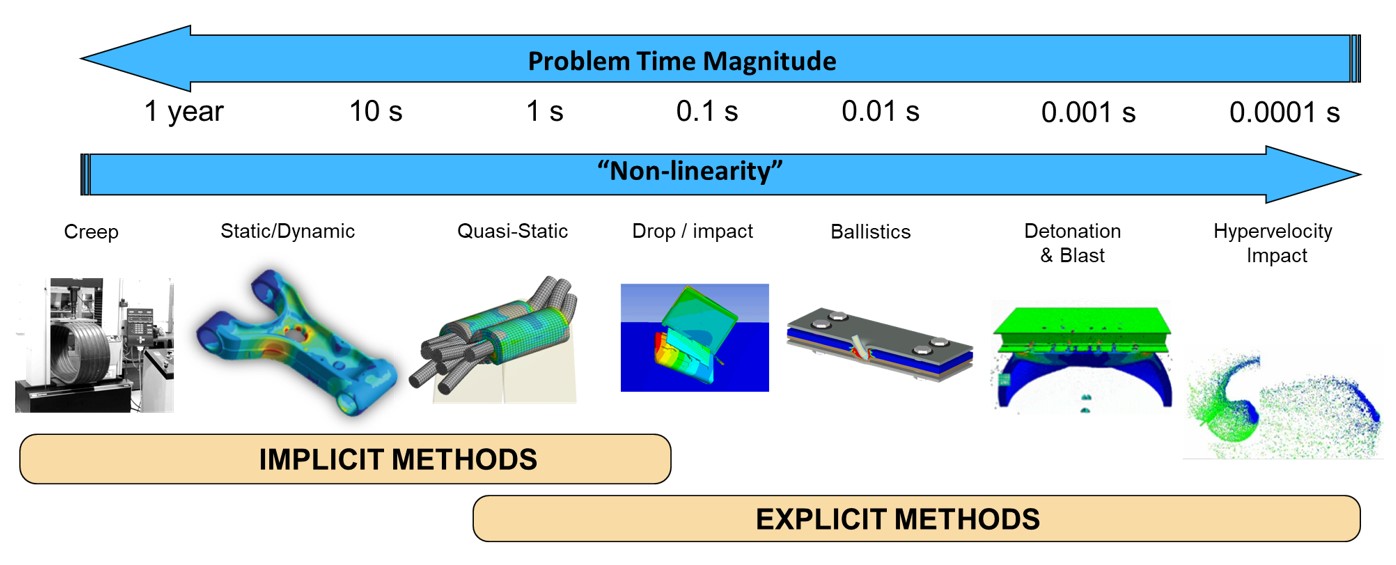
参考资源链接:[LS-PrePost:高级前处理与后处理全面教程](https://wenku.csdn.net/doc/22ae10d9h1?spm=1055.2635.3001.10343)
# 1. LS-PrePost优化设计方法概述
本章我们将简要介绍LS-PrePost优化设计方法
排序算法实现优化全攻略:J750编程性能提升秘籍
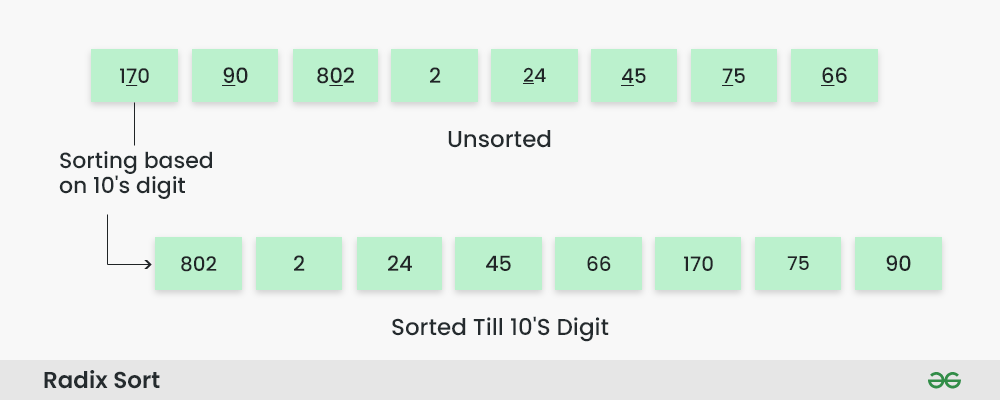
参考资源链接:[泰瑞达J750设备编程基础教程](https://wenku.csdn.net/doc/6412b472be7fbd1778d3f9e1?spm=1055.2635.3001.10343)
# 1. 排序算法基础概述
## 1.1 排序算法的重要性
在计算机科学领域,排序算法是研究数据组织、管理和检索的基础。它在数据库、操作系统、网络、信息检索等方面都
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送1年
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )