深入GROMACS力场:最佳选择与校准技巧大公开
发布时间: 2024-12-01 09:55:32 阅读量: 154 订阅数: 30 

gromacs拉伸分子动力学模拟学习笔记之mdp文件-charmm36-2022力场

参考资源链接:[Gromacs模拟教程:从pdb到gro,top文件生成及初步模拟](https://wenku.csdn.net/doc/2d8k99rejq?spm=1055.2635.3001.10343)
# 1. GROMACS力场基础与选择
分子动力学模拟在生物物理学和材料科学领域发挥着重要作用,而力场的选择和理解是进行准确模拟的关键。GROMACS作为一个广泛使用的模拟软件包,支持多种力场,为用户提供了灵活的选择。
## 1.1 力场的基本概念
力场是指在分子动力学模拟中,用于描述分子间和分子内原子间相互作用的一组数学方程。它包括原子类型、键合相互作用、角度势能、非键相互作用等元素。力场的选择直接影响模拟的准确性和可信度。
## 1.2 力场选择的重要性
正确的力场选择可以保证模拟系统达到能量最小化,更准确地反映分子的动态行为。不同的力场适用于不同类型的体系,如蛋白质、核酸或有机小分子,因此了解每种力场的特点对于模拟至关重要。
## 1.3 如何选择合适的力场
选择合适的力场需考虑模拟对象的化学性质、所需的模拟精度、计算资源的限制等因素。例如,对于蛋白质体系,常用的有AMBER、CHARMM和OPLS等力场,它们各自在参数化和经验方面有不同侧重。
在下一章中,我们将深入探讨力场参数的理论基础,为理解力场选择提供更深层次的知识支撑。
# 2. 力场参数的理论基础
## 2.1 力场的概念及其组成
### 2.1.1 原子类型和相互作用
在分子动力学模拟中,力场是一系列数学方程,用于描述分子间和分子内的原子之间的相互作用力。理解原子类型和相互作用对于构建一个可靠的力场至关重要。原子类型不仅取决于原子核和电子的组成,也包括它们可能存在的化学环境和相互作用的特殊性。例如,在蛋白质的力场中,原子被划分为不同的类型,比如碳、氢、氮、氧和硫,并根据它们是处于蛋白质的侧链还是主链来分配不同的参数。
相互作用主要包括了键合相互作用(键长、键角和二面角)和非键合相互作用(范德华力和库仑力)。键合相互作用涉及原子间通过共价键连接的直接相互作用,而非键合相互作用则包括了原子间通过空间相互作用,如氢键和疏水作用。了解这些相互作用的本质,对于建立准确的力场方程至关重要。
```mermaid
graph TD
A[原子类型] --> B[共价键]
A --> C[非共价作用]
B --> D[键长相互作用]
B --> E[键角相互作用]
B --> F[二面角相互作用]
C --> G[范德华力]
C --> H[库仑力]
```
### 2.1.2 势能函数和力场方程
势能函数用于描述分子体系内部的能量分布,是力场方程的核心组成部分。在经典力场中,势能函数通常由几个主要部分组成,包括键合相互作用的势能项(E_bond),键角的势能项(E_angle),二面角的势能项(E_dihedral),以及非键合相互作用的势能项(E_nonbonded)。力场方程可以表述为总势能(E_total)的集合:
E_total = E_bond + E_angle + E_dihedral + E_nonbonded
非键合相互作用的势能项通常由库仑项和Lennard-Jones势能项组成,分别描述了原子间由于电荷和范德华力引起的相互作用。库仑项描述了带电荷原子之间的电荷相互作用,而Lennard-Jones势能项则描述了原子之间由于距离变化而产生的吸引和排斥作用。
## 2.2 力场的分类及其适用性
### 2.2.1 经典力场与量子力场的区别
经典力场主要基于牛顿力学原理,将原子视为球体,并考虑原子核和电子的平均效果。经典力场主要包含键合和非键合相互作用,其中原子间作用力通常通过经验公式来描述。相反,量子力场采用量子力学原理来描述电子的行为和分子间的相互作用,通常更加复杂且计算成本更高。
经典力场的适用性在于其计算速度相对较快,并且对参数化良好的系统,例如有机分子和生物大分子,能提供较好的结果。但是经典力场无法描述电子的量子效应,例如共轭、电子迁移和极化等。量子力场则在处理电子相关问题,如化学反应、光谱模拟和极化效应时更有优势。
### 2.2.2 常见力场类型介绍(如AMBER, CHARMM, OPLS等)
不同类型的力场适用于不同的研究领域和分子系统。AMBER(Assisted Model Building with Energy Refinement)是一种广泛用于蛋白质和核酸的力场。AMBER力场参数化过程中考虑了氢键和离子键的特殊性,适用于模拟生物大分子的构象变化和动态行为。
CHARMM(Chemistry at HARvard Macromolecular Mechanics)力场是另一个用于蛋白质、核酸、多糖和小分子的广泛使用的力场。它支持包含溶剂和离子的复杂体系,并且能够处理各种生化过程。
OPLS(Optimized Potentials

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
GROMACS模拟流程专栏深入探讨了GROMACS模拟的各个方面,从基础概念到高级技术。专栏包含一系列文章,涵盖以下主题:
* 提升模拟速度的硬件和参数优化技巧
* 处理复杂系统的进阶指南
* 性能瓶颈和高效优化策略
* 轨迹文件解读和分析秘籍
* 并行计算的应用技巧
* 能量最小化原理和步骤
* 温度和压力控制原理和技巧
* 溶剂模型选择和优化技巧
* 质心限制技术的应用和释放
* 周期性边界条件的解析
* 反应场方法的理论和应用
* 非键相互作用的截断和长程修正技巧
* 约束算法的选择和优化
* 恒定体积和恒定压力的比较和选择
* 自由能计算的方法和技巧
该专栏旨在为GROMACS用户提供全面的指南,帮助他们优化模拟流程,解决复杂问题,并获得准确可靠的结果。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【WPF与Modbus通信】:C#新手必学的串口通讯入门秘籍(附实战项目搭建指南)
# 摘要
本文旨在探讨WPF(Windows Presentation Foundation)与Modbus通信协议的集成应用。第一章概述了WPF与Modbus通信的背景与重要性。第二章详细介绍了WPF的基础知识、界面设计、数据绑定技术及其项目结构管理。第三章则深入解析了Modbus协议的原理、通信实现方式及常见问题。在第四章,本文着重讲述了如何在WPF应用中集成Modbus通信,包括客户端与服务器的搭建和测试,以及通信模块在实战项目中的应用。最后一章提供了实战项目的搭建指南,包括需求分析、系统架构设计,以及项目实施过程的回顾和问题解决策略。通过本研究,旨在为开发人员提供一套完整的WPF与Mo
随波逐流工具深度解析:CTF编码解码的高级技能攻略(专家级教程)
# 摘要
本文全面探讨了CTF(Capture The Flag)中的编码解码技术基础与高级策略。首先介绍了编码解码的基本概念和机制,阐述了它们在CTF比赛中的应用和重要性,以及编码解码技能在其他领域的广泛使用。接着,本文深入解析了常见编码方法,并分享了高级编码技术应用与自动化处理的技巧。第三章讲述了编码算法的数学原理,探索了新思路和在信息安全中的角色。最后一章探讨了自定义编码解码工具的开发和提高解码效率的实践,以及设计复杂挑战和验证工具效果的实战演练。
# 关键字
CTF;编码解码;编码算法;信息安全;自动化处理;工具开发
参考资源链接:[随波逐流CTF编码工具:一站式加密解密解决方案]
银河麒麟V10系统与飞腾CPU的交云编译Qt5.15入门指南

# 摘要
本论文深入探讨了银河麒麟V10系统与飞腾CPU结合使用Qt5.15框架进行交叉编译的过程及其实践应用。首先概述了银河麒麟V10系统架构和飞腾CPU的技术规格,并详细介绍了Qt5.15框架的基础知识和环境搭建。随后,本论文详细阐述了Qt5.15应用开发的基础实践,包括Qt Creator的使用、信号与槽机制以及常用控件与界面布局的实现。接着,文章重
【性能提升秘诀】:5种方法加速SUMMA算法在GPU上的执行
# 摘要
本文首先概述了性能优化的理论基础和SUMMA算法原理。随后,详细介绍了基础优化技巧以及SUMMA算法在GPU上的高效实现策略,并通过性能基准测试展示了优化效果。进一步地,本文探讨了数据局部性优化和内存访问模式,以及如何通过分布式计算框架和负载均衡技术提升并行算法的效率。此外,还着重分析了GPU算力优化技巧与创新技术的应用。最后,通过实际案例分析,展示了SUMMA算法在不同领域的成功应用,并对算法的未来发展趋势及研究方向进行了展望。
# 关键字
性能优化;SUMMA算法;GPU并行计算;内存访问模式;负载均衡;算力优化;创新技术应用
参考资源链接:[矩阵乘法的并行实现-summa算
双闭环控制方法在数字电源中的应用:案例研究与实操技巧
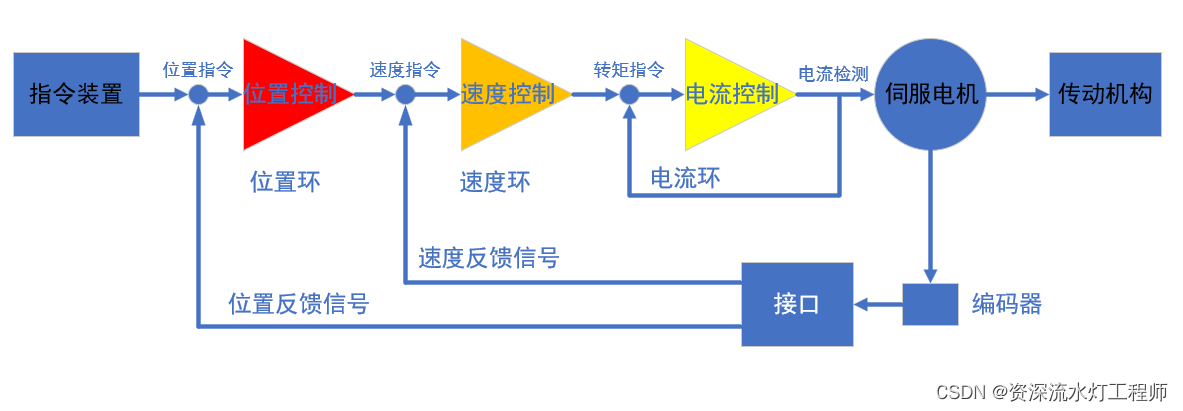
# 摘要
本文全面介绍了双闭环控制方法在数字电源中的应用,阐述了其理论基础、实现以及优化技术。首先概述了双闭环控制方法及其在数字电源工作原理中的重要性,随后详细探讨了数字电源的硬件实现与双闭环控制算法的软件实现。此外,文章还提供了实际案例分析,以展示双闭环控制在数字电源中的实现和优化过程。最后,本文展望了双闭环控制技术的未来发展趋势,包括智能控制技术的融合、创新应用以及行业标准和规范的发展。
# 关键字
双闭环控制;数字电源
Armv7-a架构深度解析:揭秘从基础到高级特性的全攻略
# 摘要
本文对ARMv7-A架构进行了全面的介绍和分析,从基础结构、高级特性到编程实践,深入探讨了该架构在现代计算中的作用。首先,概述了ARMv7-A的架构组成,包括处理器核心组件、内存管理单元和系统控制协处理器。接着,详细解读了执行状态、指令集、中断与异常处理等基础结构元素。在高级特性部分,文中重点分析了TrustZone安全扩展、虚拟化支持和通用性能增强技术。此外,还探讨了ARMv7-A在编程实践中的应用,包括汇编语言编程、操作系统支持及调试与性能分析。最后,通过应用案例,展望了ARMv7-A在未来嵌入式系统和物联网中的应用前景,以及向ARMv8架构的迁移策略。
# 关键字
ARMv7
Desigo CC高级配置案例:借鉴成功项目提升配置策略与效果

# 摘要
本文全面概述了Desigo CC在智能建筑中的应用和高级配置技术。首先介绍了Desigo CC的基本概念及其在智能建筑中的作用,接着深入探讨了配置策略的设计原理、系统要求以及从理论到实践的转化过程。文章通过实践案例分析,详细阐述了配置策略的实施步骤、问题诊断及解决方案,并对配置效果进行了评估。进一步,本文探讨了配置策略进阶技术,包括自动化配置、数据驱动优化以及安全与性能的动态平衡。最后,总结了配置过程中的经验和教训,并对
【LMS系统测试入门必读】:快速掌握操作指南与基础配置
# 摘要
本文全面介绍了学习管理系统(LMS)的测试流程,从测试的理论基础到实际的测试实践,包括系统架构解析、测试环境搭建、功能测试、性能测试以及测试自动化与持续集成。文章强调了LMS系统测试的重要性,阐述了其在软件开发生命周期中的作用,探讨了不同测试类型和方法论,以及如何进行有效的测试环境配置和数据准备。此外,本文还涉及了功能测试和性能测试的规划、执行和缺陷管理,并提出性能优化建议。最后,针对提高测试效率和质量,探讨了自动化测试框架的选择、脚本编写维护,以及持续集成的实施与管理策略。
# 关键字
学习管理系统(LMS);系统架构;性能测试;功能测试;测试自动化;持续集成
参考资源链接:[
【M-BUS主站安全防护攻略】:防雷与ESD设计的实践与心得
# 摘要
随着智能计量技术的广泛应用,M-BUS主站的安全防护已成为行业关注焦点。本文综合分析了M-BUS主站面临的雷电和静电放电(ESD)威胁,并提出了相应的防护措施。从防雷设计的基础理论出发,探讨了防雷系统层级结构、常用器件和材料,以及实施步骤中的注意事项。接着,详细阐述了ESD的物理原理、对电子设备的危害、防护策略和测试评估方法。文章进一步提出结合防雷和ESD的综合防护方案,包括设计原则、防护措施整合优化,以及案例分析。此外,还探讨了防护设备的维护、升级策略以及行业应用案例,为M-BUS主站的安全防护提供了全面的解决方案,并对行业发展趋势进行了展望。
# 关键字
M-BUS主站;安全防
稳定性保障:诺威达K2001-NWD固件兼容性测试与系统优化

# 摘要
本文详细论述了诺威达K2001-NWD固件的概述、兼容性测试理论基础、固件兼容性测试实践、系统优化理论与方法,以及诺威达K2001-NWD系统优化的实战应用。在兼容性测试部分,阐述了兼容性测试的定义、必要性分析以及测试环境的搭建
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )