GROMACS模拟:恒定体积与恒定压力的比较与选择
发布时间: 2024-12-01 11:07:21 阅读量: 37 订阅数: 41 

参考资源链接:[Gromacs模拟教程:从pdb到gro,top文件生成及初步模拟](https://wenku.csdn.net/doc/2d8k99rejq?spm=1055.2635.3001.10343)
# 1. GROMACS模拟基础介绍
## 1.1 GROMACS的定义与应用
GROMACS是一款广泛应用于生物分子模拟的软件,特别擅长处理蛋白质、核酸等大分子的模拟。GROMACS以其出色的性能和用户友好的界面在科研界享有盛誉。其模拟的主要目的包括但不限于理解分子间的相互作用、预测蛋白质的三维结构等。
## 1.2 模拟的基本步骤
GROMACS模拟的基本步骤包括:准备蛋白质结构文件、定义力场、构建溶剂模型、能量最小化、平衡体系、生产模拟运行。每个步骤都至关重要,关系到最终模拟结果的准确性。
## 1.3 模拟的输出分析
完成模拟后,需要对输出数据进行详细分析。这包括检查能量最小化是否成功、系统是否达到热力学平衡、模拟结果是否符合预期等。使用GROMACS自带工具和第三方分析软件可以帮助我们更深入地了解模拟数据。
以上内容对GROMACS模拟进行了初步的介绍,为后续章节深入探讨恒定体积与恒定压力的模拟理论和实践操作打下了基础。
# 2. 恒定体积与恒定压力模拟的理论基础
## 2.1 分子动力学模拟概述
### 2.1.1 分子动力学的基本原理
分子动力学模拟是通过数值积分解牛顿运动方程来追踪分子在空间和时间中的运动。基本原理基于牛顿第二定律,即力等于质量乘以加速度(F=ma)。在分子尺度上,力通常是通过势能函数(或称为势能表面)计算得到的,势能函数描述了原子或分子之间的相互作用。
在分子动力学模拟中,系统的总能量(动能和势能之和)通常被视为守恒量,这意味着在封闭系统中没有能量交换的情况下,系统的总能量是恒定的。模拟过程需要对时间步长进行选择,因为分子在皮秒或飞秒量级的时间尺度上会迅速移动。计算出在每个时间步长内的位置和速度,可以迭代预测下一时间步长的系统状态。
### 2.1.2 分子间相互作用与势能函数
分子间相互作用对模拟结果至关重要,它们通过势能函数来描述。势能函数包括键的伸缩、角度的弯曲、二面角的扭转以及非键相互作用(包括范德华力和库仑相互作用)。不同类型的分子和原子之间的相互作用力需要通过不同类型的势能函数来描述。
例如,蛋白质和核酸常用 CHARMM (Chemistry at HARvard Macromolecular Mechanics) 和 AMBER (Assisted Model Building with Energy Refinement) 势能函数。在无机材料模拟中,可能会使用 EAM (Embedded-Atom Method) 或 Tersoff 势能。正确的选择和调整势能函数参数对于获得准确的模拟结果至关重要。
## 2.2 恒定体积与恒定压力的理论差异
### 2.2.1 系综的概念及其意义
系综是在统计力学中,用来描述一组处于平衡状态的系统的集合。在模拟中,系综的选择决定了模拟所遵循的热力学规律。恒定体积(NVT)系综和恒定压力(NPT)系综是两个常见的选择。
在NVT系综中,模拟盒子的体积保持不变,因此,系统的压力会随着温度的变化而变化。这使得模拟适用于研究在恒定体积条件下物理性质的变化。NVT系综通常用于研究生物分子的折叠、扩散行为等在恒定体积条件下的性质。
NPT系综中,模拟盒子的体积可以变化,以保持系统压力恒定。它模拟的是在恒定压力条件下的自然环境,因此更适合研究材料的压缩性、膨胀性等性质。NPT系综对生物分子和材料科学领域都非常重要。
### 2.2.2 恒定体积与恒定压力的数学表达
恒定体积与恒定压力模拟在数学表达上的主要差异在于它们所遵循的统计力学系综。在NVT系综中,系统遵循的分布是正则系综(canonical ensemble),系统的温度、体积和粒子数是固定的,而系统的压力是变量。
在NPT系综中,系统遵循的是等温等压系综(isothermal-isobaric ensemble),系统会通过调整体积来维持恒定的压力。通过外部压力控制系统的体积变化,可以确保系统在给定的温度和压力下进行模拟。
NVT系综的模拟通常使用的是Berendsen热浴和压力耦合算法。而在NPT系综中,压力耦合算法需要能够响应体积的变化,例如 Parrinello-Rahman 或 Martyna-Tobias-Klein 算法。这些算法通过引入外部压力或压力张量,以及对盒子体积和形状进行调整,以达到热力学上的平衡状态。
# 3. 恒定体积模拟的实践操作
## 3.1 恒定体积模拟的设置
在恒定体积模拟中,系统保持固定的体积不变,研究在这样的条件下物质的热力学和动力学行为。我们通过定义模拟盒子和初始化参数,以及配置模拟运行所需的各项参数,来为恒定体积模拟做准备。
### 3.1.1 模拟盒子的定义和初始化
分子动力学模拟盒子,即模拟的计算区域,是进行恒定体积模拟的基础。盒子的尺寸和形状会直接影响模拟结果,特别是对长程相互作用的影响。
在GROMACS中定义模拟盒子的步骤大致如下:
```bash
gmx editconf -f input.gro -o box.gro -bt dodecahedron -d 1.0
```
这里,`-f input.gro` 指定了输入文件,`-o box.gro` 表示输出文件,`-bt dodecahedron` 为设置盒子类型为十二面体,而`-d 1.0` 则是设置盒子与分子间的最小距离。执行此命令后,系统会创建一个新文件`box.gro`,其中包含了定义好的模拟盒子。
### 3.1.2 模拟参数的配置
完成盒子定义后,接下来是配置模拟参数。这包括了时间步长、总模拟时间、温度和压力控制方法等。GROMACS中,这些参数被记录在`.mdp`文件中。以下是一个`.mdp`文件片段的示例:
```plaintext
; VARIOUS PREPROCESSING OPTIONS
cpp = /lib/cpp
; Simulation control options
integrator = md
dt = 0.002 ; ps
nsteps = 50000 ; total 100 ps.
comm-mode = Linear
comm-grps = system
; Output control
nstxout = 5000
nstvout = 5000
nstenergy = 5000
nstlog = 5000
; Neighborsearching
cutoff-scheme = Verlet
ns_type =
```

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
GROMACS模拟流程专栏深入探讨了GROMACS模拟的各个方面,从基础概念到高级技术。专栏包含一系列文章,涵盖以下主题:
* 提升模拟速度的硬件和参数优化技巧
* 处理复杂系统的进阶指南
* 性能瓶颈和高效优化策略
* 轨迹文件解读和分析秘籍
* 并行计算的应用技巧
* 能量最小化原理和步骤
* 温度和压力控制原理和技巧
* 溶剂模型选择和优化技巧
* 质心限制技术的应用和释放
* 周期性边界条件的解析
* 反应场方法的理论和应用
* 非键相互作用的截断和长程修正技巧
* 约束算法的选择和优化
* 恒定体积和恒定压力的比较和选择
* 自由能计算的方法和技巧
该专栏旨在为GROMACS用户提供全面的指南,帮助他们优化模拟流程,解决复杂问题,并获得准确可靠的结果。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【IBM X230主板维修宝典】:故障诊断与解决策略大揭秘
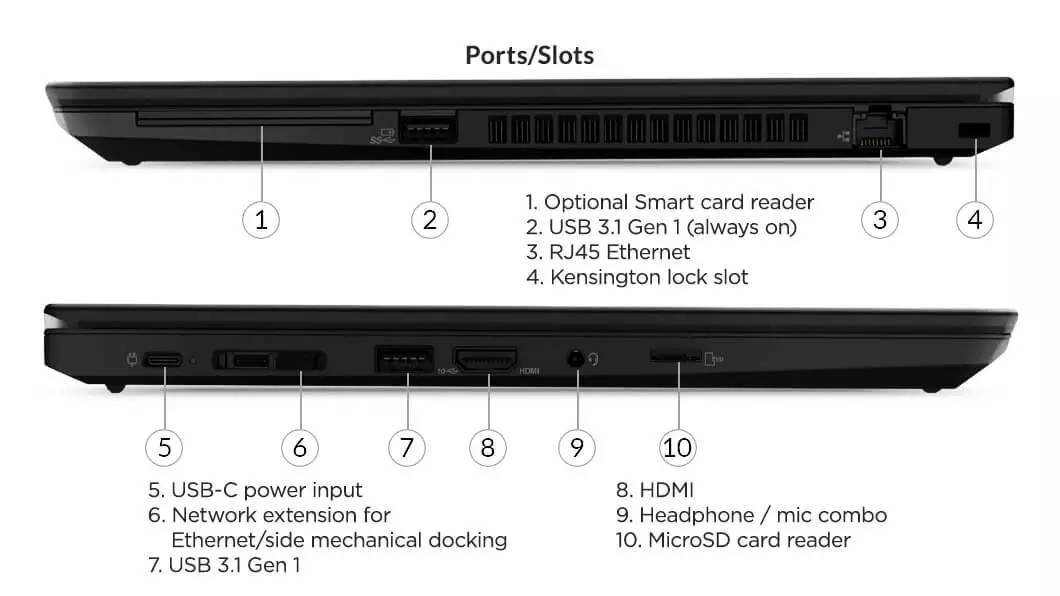
# 摘要
本文旨在全面探讨IBM X230主板的结构、故障诊断、检测与修复技巧。首先,概述了IBM X230主板的基本组成与基础故障诊断方法。随后,深入解析了主板的关键组件,如CPU插槽、内存插槽、BIOS与CMOS的功能,以及电源管理的故障分析。此外,本文详细介绍了使用硬件检测工具进行故障检测的技巧,以及在焊接技术和电子元件识别与更换过程中需要遵循的注意事项。通过对维修案例的分析,文章揭示了
ELM327中文说明书深度解析:从入门到精通的实践指南
# 摘要
ELM327设备是一种广泛应用于汽车诊断和通讯领域的接口设备,本文首先介绍了ELM327的基本概念和连接方法,随后深入探讨了其基础通信协议,包括OBD-II标准解读和与车辆的通信原理。接着,本文提供了ELM327命令行使用的详细指南,包括命令集、数据流监测与分析以及编程接口和第三方软件集成。在高级应用实践章节中,讨论了自定义脚本、安全性能优化以及扩展功能开发。最后,文章展望了ELM327的未来发展趋势,特别是在无线技术和智能汽车时代中的潜在应用与角色转变。
# 关键字
ELM327;OBD-II标准;数据通信;故障诊断;安全性能;智能网联汽车
参考资源链接:[ELM327 OBD
QNX任务调度机制揭秘:掌握这些实践,让你的应用性能翻倍

# 摘要
本文详细探讨了QNX操作系统中任务调度机制的理论基础和实践应用,并提出了一些高级技巧和未来趋势。首先概述了QNX任务调度机制,并介绍了QNX操作系统的背景与特点,以及实时操作系统的基本概念。其次,核心原理章节深入分析了任务调度的目的、要求、策略和算法,以及任务优先级与调度器行为的关系。实践应用章
CANOE工具高效使用技巧:日志截取与分析的5大秘籍
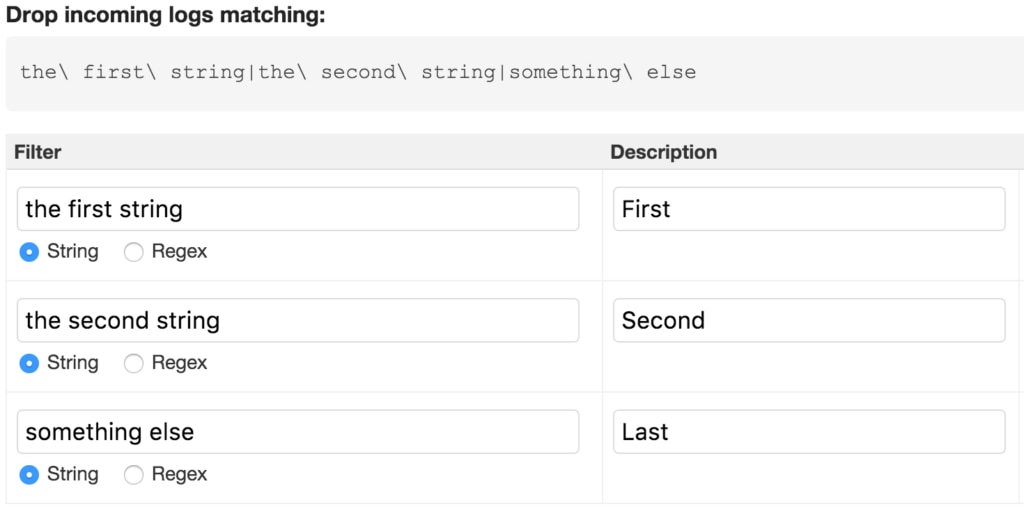
# 摘要
本文旨在提供对CANoe工具的全面介绍,包括基础使用、配置、界面定制、日志分析和高级应用等方面。文章首先概述了CANoe工具的基本概念和日志分析基础,接着详细阐述了如何进行CANoe的配置和界面定制,使用户能够根据自身需求优化工作环境。文章第三章介绍了CANoe在日志截取方面的高级技巧,包括配置、分析和问题解决方法。第四章探讨了CANoe在不同场景下的应用
【面向对象设计核心解密】:图书管理系统类图构建完全手册

# 摘要
面向对象设计是软件工程的核心方法之一,它通过封装、继承和多态等基本特征,以及一系列设计原则,如单一职责原则和开闭原则,支持系统的可扩展性和复用性。本文首先回顾了面向对象设计的基础概念,接着通过图书管理系统的案例,详细分析了面向对象分析与类图构建的实践步骤,包括类图的绘制、优化以及高级主题的应用。文中还探讨了类图构建中的高级技巧,如抽象化、泛化、关联和依赖的处理,以及约束和注释的应用。此外,本文将类图应用于图书管理
零基础到专家:一步步构建软件需求规格说明

# 摘要
软件需求规格说明是软件工程中的基
【操作系统电梯调度算法】:揭秘性能提升的10大策略和实现

# 摘要
电梯调度算法作为智能建筑物中不可或缺的部分,其效率直接影响乘客的等待时间和系统的运行效率。本文首先探讨了电梯调度算法的基础理论,包括性能指标和不同调度策略的分类。随后,文章对实现基础和进阶电梯调度算法的实践应用进行了详细介绍,包括算法编码、优化策略及测试评估方法。进一
NAND Flash固件开发必读:专家级别的4个关键开发要点
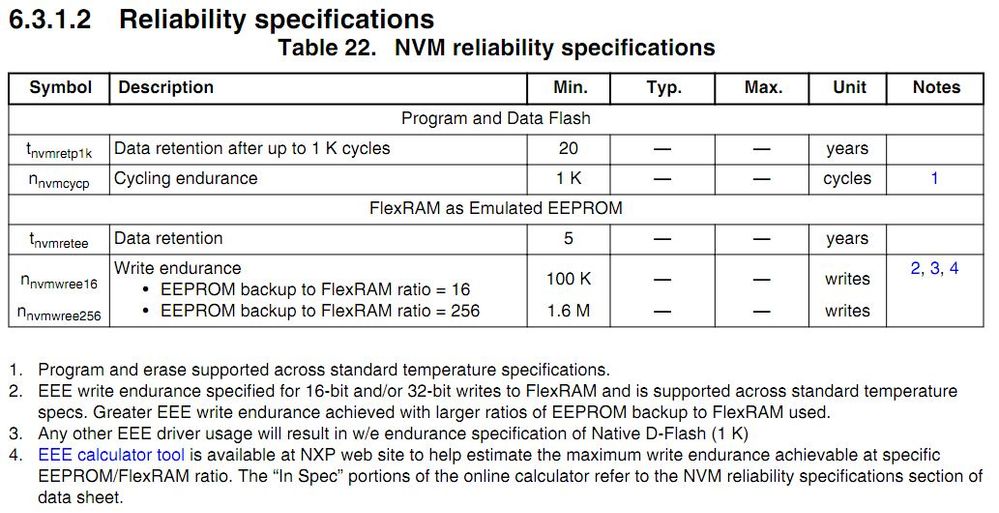
# 摘要
NAND Flash固件开发是存储技术中的关键环节,直接影响存储设备的性能和可靠性。本文首先概述了NAND Flash固件开发的基础知识,然后深入分析了NAND Flash的存储原理和接口协议。特别关注了固件开发中的错误处理、数据保护、性能优化及高级功能实现。本文通过详细探讨编程算法优化、读写效率提升
【SSD技术奥秘】:掌握JESD219A-01标准的10个关键策略

# 摘要
本论文全面概述了固态驱动器(SSD)技术,并深入探讨了JESD219A-01标准的细节,包括其形成背景、目的、影响、关键性能指标及测试方法。文章还详细讲解了SSD的关键技术要素,例如NAND闪存技术基础、SSD控制器的作用与优化、以及闪存管理技术。通过分析标准化的SSD设计与测试,本文提供了实践应用案例,同时针对JESD219A-01标准面临的挑战,提出了相应的
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )