自由能计算在GROMACS模拟中的方法与技巧:专家分享
发布时间: 2024-12-01 11:11:30 阅读量: 57 订阅数: 30 

ProtoCaller:GROMACS中相对蛋白质-配体结合自由能的全自动计算
参考资源链接:[Gromacs模拟教程:从pdb到gro,top文件生成及初步模拟](https://wenku.csdn.net/doc/2d8k99rejq?spm=1055.2635.3001.10343)
# 1. GROMACS模拟简介与安装配置
## 1.1 GROMACS模拟简介
GROMACS(GROningen MAchine for Chemical Simulations)是一款高性能的分子动力学软件,主要用于生物物理领域中的原子级模拟。GROMACS以其高效的计算性能、丰富的模拟功能和用户友好的操作界面,在学术界和工业界广泛使用。
GROMACS能够模拟从简单的原子系统到复杂的生物大分子,如蛋白质、核酸、脂质、碳水化合物等,以及它们在水或其他溶剂中的行为。GROMACS在蛋白质折叠、药物设计、生物材料性质研究等方面均有应用。
## 1.2 GROMACS的安装配置
GROMACS的安装方式多样,可选择从源代码编译安装,也可通过预编译的二进制包或通过包管理器安装。以下是基于Linux系统,使用包管理器安装GROMACS的简单步骤:
1. 更新系统包列表:
```bash
sudo apt-get update
```
2. 安装GROMACS:
```bash
sudo apt-get install gromacs
```
3. 验证安装是否成功:
```bash
gmx -version
```
请注意,对于特定版本的GROMACS或在非Linux系统上安装,可能需要不同的安装方法。详细安装指南可以在GROMACS官方网站找到。安装完成后,还需要配置环境变量,设置GROMACS工具的路径,以便在任何位置调用它们。安装和配置完成后,就可以开始探索GROMACS模拟世界了。
# 2. 自由能计算理论基础
## 2.1 分子动力学模拟与自由能
### 2.1.1 自由能的定义与热力学原理
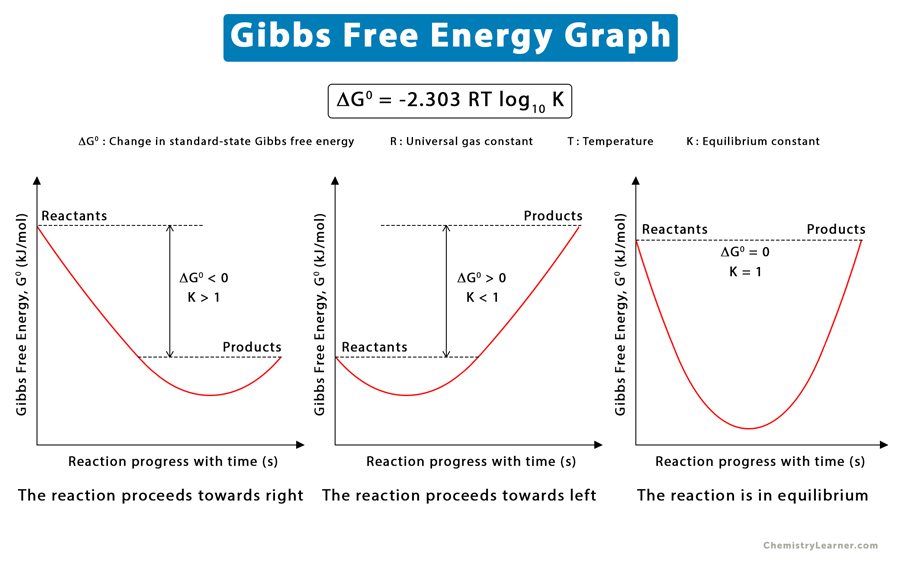
自由能,通常指的是亥姆霍兹自由能(A)或吉布斯自由能(G),是热力学中描述系统状态的一个重要函数。在恒温恒压条件下,系统趋向于自由能最小化的状态。对于化学反应、相变等过程,自由能差ΔG是决定其自发性与平衡位置的关键因素。
亥姆霍兹自由能是指系统在恒温条件下,可用来做功的能量,它等于内能与熵乘以温度的乘积(A=U-TS)。而在实际操作中,分子动力学模拟通常是在恒容条件下进行,此时考虑的是吉布斯自由能,它定义为系统的内能加上压力与体积的乘积再减去温度与熵的乘积(G=U+PV-TS)。
热力学第一定律告诉我们能量守恒,即系统内能的变化等于外界对系统做的功与系统传递给外界的热量之和。自由能的物理意义在于它能够衡量在恒温条件下系统可做多少功。在生物大分子的模拟中,理解分子在不同状态下的自由能变化对于研究蛋白质折叠、酶催化和药物结合等复杂生物过程具有至关重要的意义。
### 2.1.2 自由能计算在模拟中的重要性
在分子动力学模拟中,通过自由能计算可以预测分子间相互作用的强度、蛋白质-配体的结合亲和力、蛋白质构象变化的倾向等。这些计算结果不仅提供了对生物分子行为的深入理解,而且在药物设计与材料科学中具有实际应用价值。
举例来说,药物分子与靶点蛋白的结合自由能决定了其亲和力大小,因此准确计算自由能对于筛选高效药物候选分子至关重要。此外,自由能计算在预测酶催化过程的机理、分析配体与蛋白结合的热力学性质等方面也有着广泛的应用。
## 2.2 自由能计算方法概述
### 2.2.1 热力学积分法(TI)
热力学积分法是通过在不同的虚拟状态(由参数λ定义)之间进行积分,计算系统的自由能变化。在分子动力学模拟中,通过定义λ空间,并对不同的λ值进行模拟,可以构建出自由能曲线。
具体地,可以通过改变原子间相互作用的强度(例如,范德华力和静电作用的强度)来定义λ点,然后分别在不同的λ值下运行模拟。通过计算不同λ点之间的自由能差,我们可以估计任意两点间的真实自由能差异。
### 2.2.2 稀释法(FEP)
稀释法是一种基于粒子插入的自由能计算方法,它通过模拟粒子的添加或删除来计算自由能变化。在分子动力学模拟中,通过逐步改变模拟体系中粒子的属性,从一个已知的状态平滑过渡到一个未知的状态,从而估计两种状态之间的自由能差异。
在实际操作中,FEP涉及在模拟过程中逐渐调整粒子的类型,而其他的物理量(如温度、体积)保持不变。通过细致的模拟步骤,FEP可以非常精确地计算出自由能变化,但是由于其对模拟的细致程度要求很高,相应的计算成本也较大。
### 2.2.3 非平衡态方法(NEB)
非平衡态方法(Nonequilibrium Switching,NEB)是一种直接测量自由能变化的方法,它通过对系统施加一个非平衡的驱动来产生功,从而计算自由能差。这种方法的特点是计算效率高,但是需要仔细设计非平衡驱动过程以确保结果的准确性。
NEB方法通常用于快速估计两个状态之间的自由能差异,它不需要对整个系统进行长时间的平衡模拟,因此在处理大规模系统时更为高效。然而,对非平衡过程的分析需要细致的物理理解和恰当的理论指导。
在下一章中,我们将探讨如何在GROMACS模拟软件中实现上述自由能计算方法,并进一步讨论实际操作中的技巧和优化。通过具体案例和深入分析,我们将深入理解自由能计算在分子动力学模拟中的应用。
# 3. GROMACS中的自由能计算实践
## 3.1 GROMACS中的自由能计算准备工作
### 3.1.1 系统的构建与参数化
在使用GROMACS进行自由能计算之前,首先需要构建好模拟系统并进行适当的参数化。模拟系统构建通常包括选择合适的力场、添加溶剂、离子化处理和能量最小化等步骤。
选择力场是构建系统的第一步。力场决定了模拟中分子的相互作用和动力学行为。常见的力场包括AMBER、CHARMM、OPLS-AA等

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
GROMACS模拟流程专栏深入探讨了GROMACS模拟的各个方面,从基础概念到高级技术。专栏包含一系列文章,涵盖以下主题:
* 提升模拟速度的硬件和参数优化技巧
* 处理复杂系统的进阶指南
* 性能瓶颈和高效优化策略
* 轨迹文件解读和分析秘籍
* 并行计算的应用技巧
* 能量最小化原理和步骤
* 温度和压力控制原理和技巧
* 溶剂模型选择和优化技巧
* 质心限制技术的应用和释放
* 周期性边界条件的解析
* 反应场方法的理论和应用
* 非键相互作用的截断和长程修正技巧
* 约束算法的选择和优化
* 恒定体积和恒定压力的比较和选择
* 自由能计算的方法和技巧
该专栏旨在为GROMACS用户提供全面的指南,帮助他们优化模拟流程,解决复杂问题,并获得准确可靠的结果。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
IQxel-M8X故障诊断宝典:无线网络故障快速解决之道
# 摘要
本文对IQxel-M8X设备进行了全面介绍,并详细阐述了无线网络的基础理论、常见问题及其故障诊断方法。通过对无线信号传播原理、802.11标准的演进、无线网络故障的分类、安全威胁及预防措施的分析,本文提供了对无线网络深入理解和故障处理的策略。此外,本文还探讨了IQxel-M8X故障诊断工具的使用、网络优化实践,以及网络监控与管理的策略。通过案例分析和故障模拟演练,本文旨在提高无线网络维护的效率和效果。最后,本文展望了无线网络技术的发展趋势和IQxel-M8X产品的未来演进,以支持无线网络领域的持续创新和发展。
# 关键字
IQxel-M8X设备;无线网络;故障诊断;网络安全;网络优
微信小程序手机号授权:深入案例分析及改进技巧
# 摘要
本文全面探讨了微信小程序手机号授权机制,从理论基础到进阶技巧,再到未来趋势进行了深入分析。首先,概述了微信小程序手机号授权的开发环境设置和授权流程,随后通过实际案例展示了授权需求分析、流程实现和常见问题解决。文章重点讨论了如何提升用户体验和保护用户隐私,并提供了高级措施和优化建议。最后,展望了微信小程序及手机号授权的未来发展方向,包括行业标准和合规性,以及行业内的最佳实践。
# 关键字
微信小程序;手机号授权;用户体验;隐私保护;授权流程;行业趋势
参考资源链接:[微信小程序:轻松获取用户手机号授权登录](https://wenku.csdn.net/doc/6412b49cbe
代码审查实战】:提升软件质量的最佳实践与策略
# 摘要
代码审查是确保软件质量、维护代码健康的重要实践。本文首先介绍了代码审查的概念及其重要性,强调了准备工作在成功实施审查过程中的核心地位,包括设定审查目标、选择工具和环境、规划流程和时间表。随后,文章深入探讨了实施代码审查的多种方法,强调了手动和自动化审查工具的互补性以及沟通与反馈的重要性。此外,本文还识别并解决了代码审查实践中遇到的挑战,并提供了改进审查流程和策略的建议。最后,文章展望了代码审查策略的未来趋势,重点是敏捷开发环境下的审查以及技术创新对审查实践的影响,同时强调了建立持续学习和改进文化的重要性。
# 关键字
代码审查;质量保证;审查工具;审查流程;敏捷开发;持续学习
参
检查发货单中的异常处理:需求分析与设计的5大策略
# 摘要
异常处理在业务流程中扮演着至关重要的角色,尤其是针对发货单的检查,它可以确保订单的准确性、及时性与合规性。本文首先介绍了异常处理的基本理论,包括异常的定义、类型及处理原则,以及发货单的关键数据结构和字段验证。随后,文章深入探讨了实践中的检查策略,涵盖手动与自动化检测方法、异常处理流程设计,以及数据分析技术在异常模式识别中的应用。通过具体实践案例的分析,本文展现了需求分析与策略设计的执行过程和效果评估。最后,本文展望了异常处理技术的未来发展,并讨论了需求分析与设计的创新方法,以及战略规划和组织调整的重要性。
# 关键字
异常处理;发货单检查;数据分析;异常检测;需求分析;流程设计
ISE仿真与测试:自动化测试脚本编写指南
# 摘要
随着集成电路设计复杂性的增加,ISE仿真与测试变得愈发重要。本文深入探讨了自动化测试脚本的理论基础、编写实践以及高级应用。文章首先概述了自动化测试脚本的目的和作用,强调了其在ISE环境下的优势。接着,详细阐述了测试脚本的基本组成、设计原则以及性能评估与优化。此外,还介绍了ISE仿真测试脚本与ISE工具的集成、数据驱动测试、并行测试和负载测试的高级应用。最后,文中通过案例研究,展示了自动化测试脚本在实际项目中的应用,分析了遇到的挑战和解决方案,为提升测试效率和质量提供了实践指导和经验分享。
# 关键字
ISE仿真;自动化测试脚本;测试用例;性能优化;数据驱动测试;并行测试
参考资源
数据不丢失:Hollysys_Macs6.5.4B2备份与恢复最佳实践
# 摘要
随着信息技术的不断进步,数据备份与恢复的重要性日益凸显,尤其是在关键业务系统中。本文详细介绍了Hollysys_Macs6.5.4B2系统的备份与恢复实践,包括基础概念、安装与配置、备份策略、数据验证、自动化实现以及高级恢复技术。文章通过系统概述、实践操作指南以及高级应用探讨,旨在为用户提供全面的数据备份与恢复解决方案,确保数据安全和业务连续性。同时,本文还探讨了故障排除、性能优化及预防性维护,以帮助用户提高备份恢复系统的运行效率和稳定性。
# 关键字
数据备份;数据恢复;Hollysys_Macs6.5.4B2;备份策略;性能优化;故障排除
参考资源链接:[解决Hollysys
组态王与PLC通信秘籍:数据交换与硬件集成详解
# 摘要
本文深入探讨了组态王与PLC通信的技术细节与实践应用,首先概述了组态王与PLC通信的理论基础和通信协议。随后详细介绍了硬件集成的实践技巧,包括前期准备、实施过程以及问题解决策略。在数据交换的高级应用部分,分析了数据处理技术、高级通信协议的应用以及数据交换的安全性措施。最后,通过案例研究,探讨了组态王与PLC集成的实践成果和面临的智能化挑战与机遇。本文旨在为自动化工程师提供一个全面的参考指南,以实现高效、安全的工业控制系统集成。
# 关键字
组态王;PLC通信;硬件集成;数据交换;通信协议;智能化集成
参考资源链接:[组态王:历史数据查询与报表制作教程](https://wenku
展锐平台下载工具性能飞跃:速度与稳定性提升指南
# 摘要
展锐平台下载工具概述为起点,本文深入探讨了通过理论基础、实践策略、技术路径,以及案例研究来提升下载速度和工具稳定性。性能提升的理论基础部分详细介绍了下载工具的工作原理,包括网络协议、下载算法效率,以及系统性能优化理论。在提升下载速度的实践策略章节中,探讨了网络连接优化、缓存与预取技术、多线程与多路下载的应用。同时,为了增强工具的稳定性,本文也讨论了容错与恢复机制、服务器负载均衡、健康检查和用户体验管理。最后,通过案例研究与实操演练展示了优化实例和性能调优技巧,并对未来的下载技术趋势和挑战进行了展望,指出P2P技术、AI应用以及安全性和兼容性问题的应对策略。
# 关键字
展锐平台;下
【仿真工具核心功能】:NS-3.17网络模拟器深度剖析,揭秘仿真的秘密
# 摘要
NS-3.17作为一款先进的网络模拟器,为研究者和开发者提供了强大的网络仿真能力。本文首先概述了NS-3.17的基础架构和核心理论,阐述了其在网络模拟与仿真技术中的应用,以及模块化组件和网络模型的设计与实现。接着,详细讨论了如何在实践中搭建和配置NS-3.17环境,以及如何进行网络协议仿真实践和性能评估。文章还介绍了一些高级功能,包括模块拓展、仿真调试优化和模拟器的可视化。最后,通过对无线传感网络、大规模网络环境以及网络安全仿真实验的案例研究,展示了NS-3.17在模拟复杂网络场景中的应用。本文旨在为读者提供一个全面了解NS-3.17的平台,并指导其在具体项目中的应用。
# 关键字
团队协作与创新:美赛E题获奖团队的经验深度分享
# 摘要
本文综合分析了美赛E题的竞争环境,系统地讨论了团队组建、创新思维培养、实战演练和经验总结等方面的重要性和策略。通过探讨如何挑选合适的队友、建立高效的沟通机制、管理团队冲突、培养和实践创新思维、以及优化模型构建等,为参赛者提供了全面的准备指南。文章还强调了团队成员间信任建设的重要性,并通过案例分析展示了创新策略在实际竞赛中的应用效果。最后,本文总结了获奖经验,并对未来可能的赛题趋势进行了预测,为参赛者提供了持续创新和团队成长的参考方向。
# 关键字
美赛E题;团队组建;创新思维;实战演练;信任建设;经验总结
参考资源链接:[光污染评估与干预策略:LSN模型与PIA-NN分析](ht
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )