MATLAB求导在生物信息学中的价值:助力生物信息学研究,探索生命奥秘
发布时间: 2024-05-23 12:34:42 阅读量: 7 订阅数: 15 

# 1. MATLAB求导的理论基础
MATLAB求导是计算函数或表达式对变量导数的过程,是数学分析和科学计算中的一项基本操作。在MATLAB中,求导可以通过使用内置函数`gradient`和`diff`实现。
`gradient`函数用于计算多变量函数的梯度,它返回一个与输入函数具有相同维度的矩阵,其中每个元素代表函数在该点沿相应变量的偏导数。
`diff`函数用于计算一维函数的差分,它返回一个与输入函数长度减一的新向量,其中每个元素代表函数在相邻点之间的差值。通过使用`diff`函数两次,可以近似计算函数的导数。
# 2. MATLAB求导在生物信息学中的应用
MATLAB求导在生物信息学中有着广泛的应用,主要体现在数据处理和模型构建两个方面。
### 2.1 生物信息学数据处理中的求导
#### 2.1.1 序列比对中的求导
在生物信息学中,序列比对是比较两个或多个序列相似性的基本技术。求导可以用于优化序列比对算法,提高比对准确性和效率。
**代码块:**
```matlab
function [score, alignment] = NeedlemanWunsch(seq1, seq2)
% 初始化得分矩阵和回溯指针矩阵
score = zeros(length(seq1) + 1, length(seq2) + 1);
pointer = zeros(length(seq1) + 1, length(seq2) + 1);
% 填充第一行和第一列
for i = 1:length(seq1) + 1
score(i, 1) = -i;
pointer(i, 1) = 2; % 指向左
end
for j = 1:length(seq2) + 1
score(1, j) = -j;
pointer(1, j) = 1; % 指向上
end
% 填充其余部分
for i = 2:length(seq1) + 1
for j = 2:length(seq2) + 1
% 计算当前位置的分数
match = score(i - 1, j - 1) + (seq1(i - 1) == seq2(j - 1));
insert = score(i - 1, j) - 1;
delete = score(i, j - 1) - 1;
% 选择最高分并更新回溯指针
[score(i, j), pointer(i, j)] = max([match, insert, delete]);
end
end
% 回溯并生成比对
alignment1 = '';
alignment2 = '';
i = length(seq1);
j = length(seq2);
while i > 0 && j > 0
switch pointer(i, j)
case 1 % 向上
alignment1 = [seq1(i - 1), alignment1];
alignment2 = ['-', alignment2];
i = i - 1;
case 2 % 向左
alignment1 = ['-', alignment1];
alignment2 = [seq2(j - 1), alignment2];
j = j - 1;
case 3 % 斜对角线
alignment1 = [seq1(i - 1), alignment1];
alignment2 = [seq2(j - 1), alignment2];
i = i - 1;
j = j - 1;
end
end
% 返回比对结果
return;
end
```
**逻辑分析:**
此代码实现了Needleman-Wunsch序列比对算法,其中求导用于计算比对过程中的得分矩阵。得分矩阵记录了每个位置的最佳比对分数,并通过回溯指针矩阵进行回溯以生成比对结果。
#### 2.1.2 基因表达分析中的求导
基因表达分析是研究基因功能和调控的重要手段。求导可以用于分析基因表达数据,识别差异表达基因和调控模式。
**代码块:**
```matlab
% 导入基因表达数据
data = importdata('gene_expression.txt');
% 对数据进行归一化
data = normalize(data, 'zscore');
% 计算基因表达量的导数
derivatives = diff(data, 1, 2);
% 识别差异表达基因
diff_genes = find(abs(derivatives) > 0.5);
```
**逻辑分析:**
此代码对基因表达数据进行归一化,然后计算每个基因表达量的导数。导数反映了基因表达量的变化率,可以用于识别差异表达基因。
### 2.2 生物信息学模型构建中的求导
#### 2.2.1 动力学模型中的求导
动力学模型用于
 最低0.47元/天 解锁专栏
最低0.47元/天 解锁专栏 赠618次下载
 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏深入探讨了 MATLAB 求导的强大功能,提供了一系列技巧和方法,帮助读者轻松解决微积分难题。从基础概念到进阶技术,专栏涵盖了 10 个实用技巧,揭示了 MATLAB 求导的奥秘。通过循序渐进的讲解和丰富的示例,读者将掌握 MATLAB 求导的精髓,提升微积分问题解决能力。无论是学生、研究人员还是工程师,本专栏都将成为 MATLAB 求导的宝贵指南,助您在微积分领域取得成功。
专栏目录
最低0.47元/天 解锁专栏
赠618次下载
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
Python生成Excel文件:开发人员指南,自动化架构设计
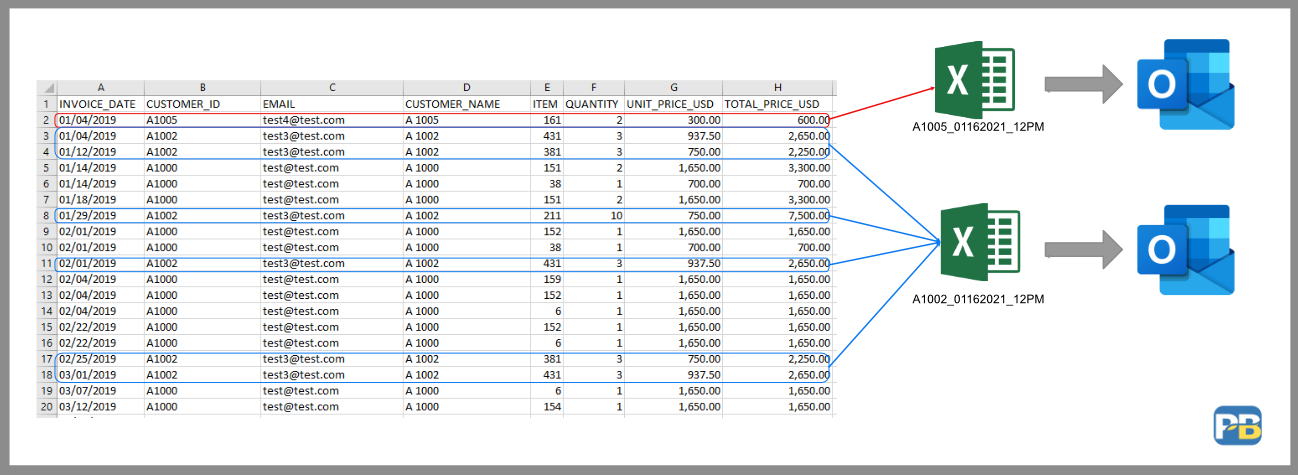
# 1. Python生成Excel文件的概述**
Python是一种功能强大的编程语言,它提供了生成和操作Excel文件的能力。本教程将引导您了解Python生成Excel文件的各个方面,从基本操作到高级应用。
Excel文件广泛用于数据存储、分析和可视化。Python可以轻松地与Excel文件交互,这使得它成为自动化任务和创建动态报表的理想选择。通过使用Python,您可以高效地创建、读取、更新和格式化E
Python变量作用域与云计算:理解变量作用域对云计算的影响

# 1. Python变量作用域基础
变量作用域是Python中一个重要的概念,它定义了变量在程序中可访问的范围。变量的作用域由其声明的位置决定。在Python中,有四种作用域:
- **局部作用域:**变量在函数或方法内声明,只在该函数或方法内可见。
- **封闭作用域:**变量在函数或方法内声明,但在其外层作用域中使用。
- **全局作用域:**变量在模块的全局作用域中声明
Python Excel读写项目管理与协作:提升团队效率,实现项目成功

# 1. Python Excel读写的基础**
Python是一种强大的编程语言,它提供了广泛的库来处理各种任务,包括Excel读写。在这章中,我们将探讨Python Excel读写的基础,包括:
* **Excel文件格式概述:**了解Excel文件格式(如.xlsx和.xls)以及它们的不同版本。
* **Python Excel库:**介绍用于Python
Python3.7.0安装与最佳实践:分享经验教训和行业标准
# 1. Python 3.7.0 安装指南
Python 3.7.0 是 Python 编程语言的一个主要版本,它带来了许多新特性和改进。要开始使用 Python 3.7.0,您需要先安装它。
本指南将逐步指导您在不同的操作系统(Windows、macOS 和 Linux)上安装 Python 3.7.0。安装过程相对简单,但根据您的操作系统可能会有所不同。
# 2. Pyt
Python字符串为空判断的自动化测试:确保代码质量

# 1. Python字符串为空判断的必要性
在Python编程中,字符串为空判断是一个至关重要的任务。空字符串表示一个不包含任何字符的字符串,在各种场景下,判断字符串是否为空至关重要。例如:
* **数据验证:**确保用户输入或从数据库中获取的数据不为空,防止程序出现异常。
* **数据处理:**在处理字符串数据时,需要区分空字符串和其他非空字符串,以进行不同的操作。
* **代码可读
Python Requests库:常见问题解答大全,解决常见疑难杂症

# 1. Python Requests库简介
Requests库是一个功能强大的Python HTTP库,用于发送HTTP请求并处理响应。它提供了简洁、易用的API,可以轻松地与Web服务和API交互。
Requests库的关键特性包括:
- **易于使用:**直观的API,使发送HTTP请求变得简单。
- **功能丰富:**支持各种HTTP方法、身份验证机制和代理设
PyCharm Python路径与移动开发:配置移动开发项目路径的指南

# 1. PyCharm Python路径概述
PyCharm是一款功能强大的Python集成开发环境(IDE),它提供
Python Lambda函数在DevOps中的作用:自动化部署和持续集成
# 1. Python Lambda函数简介**
Lambda函数是一种无服务器计算服务,它允许开发者在无需管理服务器的情况下运行代码。Lambda函数使用按需付费的定价模型,只在代码执行时收费。
Lambda函数使用Python编程语言编写
Jupyter Notebook安装与配置:云平台详解,弹性部署,按需付费

# 1. Jupyter Notebook概述**
Jupyter Notebook是一个基于Web的交互式开发环境,用于数据科学、机器学习和Web开发。它提供了一个交互式界面,允许用户创建和执行代码块(称为单元格),并查看结果。
Jupyter Notebook的主
Python连接SQL Server连接池与并发:处理高并发连接
# 1. Python连接SQL Server连接池**
### 1.1 连接池的概念和优势
连接池是一种用于管理数据库连接的机制,它在内存中维护一个预先建立的连接池。当应用程序需要连接数据库时,它可以从连接池中获取一个可用的连接,而无需重新建立连接。
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
赠618次下载
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )