初识LAMMPS: 强大的分子动力学模拟软件简介
发布时间: 2024-03-27 20:00:08 阅读量: 216 订阅数: 41 
# 1. 介绍分子动力学模拟与LAMMPS
## 1.1 什么是分子动力学模拟?
分子动力学模拟是一种计算方法,通过在计算机上模拟原子和分子之间的相互作用,来研究物质的结构和性质变化。借助此方法,可以模拟原子和分子在不同条件下的运动轨迹,探究物质的宏观性质。
## 1.2 LAMMPS软件概述
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一款开源的分子动力学软件,适用于模拟原子、离子和分子等微观粒子的运动行为。由Sandia国家实验室开发,支持多种粒子间的相互作用力场,具有高效、可扩展性强的特点。
## 1.3 分子动力学模拟在科学研究中的应用
分子动力学模拟广泛应用于材料科学、生物医药、化学等领域。通过模拟原子之间的相互作用,可以揭示物质的动力学过程和结构性质,帮助科研人员理解分子尺度下的现象和机制。
# 2. LAMMPS软件的基础知识
分子动力学模拟作为一种重要的计算方法,在许多领域得到广泛应用。而LAMMPS作为一款开源的分子动力学模拟软件,在其独特的特点和优势下备受科研工作者的青睐。
### 2.1 LAMMPS的特点和优势
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)的独特之处在于其高度并行化的设计,使其能够处理大规模的分子系统,同时具备较高的计算效率。其优势包括但不限于:
- **可扩展性:** LAMMPS支持多种作业系统和硬件平台,可以在单个CPU到大规模并行计算机集群中运行,满足不同计算需求。
- **模拟能力:** LAMMPS内置了多种模拟算法和相互作用力场,可以模拟不同类型的材料和体系,包括原子、分子、粒子和连续介质等。
- **灵活性:** 用户可以通过编写自定义脚本和插件来扩展LAMMPS的功能,满足特定研究需求。
### 2.2 LAMMPS的安装与配置
要使用LAMMPS进行分子动力学模拟,首先需要安装和配置软件环境。一般而言,可以按照以下步骤进行:
1. **下载安装:** 访问LAMMPS官方网站(https://lammps.sandia.gov/)下载最新版本的软件包,并按照官方提供的安装说明进行安装。
2. **编译配置:** 在安装完成后,根据所需的模块和功能,选择相应的编译选项进行配置,并进行编译生成可执行文件。
3. **验证测试:** 在安装完成后,可以通过运行LAMMPS提供的测试案例来验证软件是否正常工作。
### 2.3 LAMMPS的基本工作原理
LAMMPS基于经典的分子动力学理论,通过数值积分的方法,模拟系统中原子或分子的运动轨迹,并根据相互作用势能计算系统的力学性质。其基本工作原理包括以下几个步骤:
1. **初始化系统:** 定义原子的初始位置、速度和相互作用势能参数。
2. **迭代求解:** 通过积分运动方程,更新原子的位置和速度,计算相互作用力。
3. **相互作用计算:** 根据所选的相互作用势,计算原子之间的相互作用力,并更新系统状态。
4. **数据输出:** 输出模拟结果,包括能量变化、结构演变、动力学性质等信息。
以上是关于LAMMPS软件的基础知识介绍,下一步将进入到搭建LAMMPS模拟环境的具体步骤。
# 3. LAMMPS模拟环境的搭建
在进行分子动力学模拟之前,需要先搭建合适的模拟环境,包括定义原子类型和相互作用势、设定模拟系统参数以及构建模拟系统结构。以下是LAMMPS模拟环境搭建的基本步骤:
#### 3.1 定义原子类型和相互作用势
在LAMMPS中,需要定义模拟系统中各种原子的类型,以及它们之间的相互作用势。这包括描述原子间的键合、斥力和相互作用能量等。用户可以根据模拟系统的特性选择不同类型的原子和相互作用势函数,如Lennard-Jones势、Coulomb势等。
```python
# 示例:定义原子类型和相互作用势
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0
pair_coeff 1 2 1.2 1.4
pair_coeff 2 2 1.4 0.8
```
#### 3.2 设定模拟系统参数
在模拟环境中,需要设定模拟系统的基本参数,如温度、压力、模拟时间步长等。这些参数将直接影响模拟过程中原子的运动轨迹和相互作用力的计算。
```python
# 示例:设定模拟系统参数
variable temp equal 300.0
variable pressure equal 1.0
timestep 0.001
```
#### 3.3 构建模拟系统结构
构建模拟系统的结构是指确定模拟系统中原子的初始位置和速度。用户可以通过随机生成或指定坐标等方式来构建初始系统结构,以便进行后续的动力学模拟计算。
```python
# 示例:构建模拟系统结构
lattice fcc 0.8
create_box 2 box
create_atoms 1 box
mass 1 1.0
mass 2 1.5
velocity all create 300.0 87287
```
通过以上步骤,我们可以构建一个完整的LAMMPS模拟环境,准备进行分子动力学模拟实验。在后续的章节中,将会详细介绍如何编写输入文件、设置模拟参数以及启动模拟过程。
# 4. 运行LAMMPS模拟
在这一章节中,我们将详细讨论如何在LAMMPS中运行分子动力学模拟,包括输入文件的编写、模拟参数设置、启动模拟以及结果输出等步骤。
### 4.1 输入文件的编写
在运行LAMMPS模拟之前,首先需要编写输入文件,通常以`.in`作为扩展名。输入文件包含了模拟系统的参数设置、计算步数、温度压力等控制信息,以及物质的初态信息等。
以下是一个简单的LAMMPS输入文件示例:
```markdown
# 设置模拟基本信息
units metal
dimension 3
boundary p p p
# 定义原子类型
atom_style atomic
atom_modify map array
# 设定计算参数
lattice fcc 3.5
region box block 0 10 0 10 0 10
create_box 1 box
create_atoms 1 box
# 设置相互作用势
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0
# 运行
thermo 100
thermo_style custom step temp
run 1000
```
### 4.2 模拟参数设置
在输入文件中,我们可以设定模拟系统的温度、压力、能量收敛标准等参数。通过调整这些参数,可以对模拟系统的行为进行精细控制。
### 4.3 启动模拟与结果输出
完成输入文件的编写后,通过在终端运行以下命令来启动LAMMPS模拟:
```bash
lmp_serial -in input_file_name.in
```
LAMMPS将按照输入文件中设定的参数和步数进行模拟运行,并在运行结束后输出相应的结果数据,如模拟过程中的能量、温度变化、原子位置等信息,以供后续分析使用。
# 5. 分子动力学模拟实例
分子动力学模拟是一种在计算机上模拟原子和分子运动的技术,通过模拟粒子间的相互作用,可以研究物质的结构、性质和变化过程。接下来,我们将通过几个实例来展示如何利用LAMMPS软件进行分子动力学模拟。
### 5.1 模拟液体分子的运动
在这个实例中,我们将模拟一组液体分子的运动。首先,我们需要定义分子类型、相互作用势等参数,并构建模拟系统。然后编写输入文件,设置模拟参数,并运行LAMMPS模拟。
```python
# Python伪代码示例
# 定义原子类型和相互作用势
atom_types = {'O': 1, 'H': 2}
interactions = {'O-O': 'lj/cut', 'O-H': 'lj/cut', 'H-H': 'lj/cut'}
# 设定模拟系统参数
box_size = 10.0
num_atoms = 100
temperature = 300
# 构建模拟系统结构
system = build_liquid_molecules(box_size, num_atoms)
# 编写输入文件
input_file = create_input_file(system, atom_types, interactions, temperature)
# 模拟参数设置
timestep = 0.001
total_steps = 1000
# 启动模拟与结果输出
run_simulation(input_file, timestep, total_steps)
```
通过这个实例,我们可以观察到液体分子在不同温度下的运动状态,了解其动力学行为。
### 5.2 研究固体结构的稳定性
在这个实例中,我们将研究固体材料的结构稳定性。通过LAMMPS模拟,我们可以在不同条件下改变原子位置或应力,观察固体结构的变化和稳定性情况。
```java
// Java示例代码
// 构建固体晶胞
Cell cell = new Cell(10.0);
Solid solid = build_solid_structure(cell);
// 添加温度和压力条件
solid.setTemperature(300);
solid.applyStress(1.0, 0.0, 0.0, 0.0, 1.0, 0.0, 0.0, 0.0, 1.0);
// 运行模拟
solid.runSimulation(1000);
```
通过这个实例,我们可以了解固体材料在外部条件作用下的结构变化和相互作用情况,有助于进一步研究固体的性质和稳定性。
### 5.3 模拟化学反应过程
在这个实例中,我们将模拟化学反应的过程。通过LAMMPS软件,可以模拟原子或分子之间的键合和断裂过程,从而研究化学反应的动力学和机制。
```javascript
// JavaScript示例代码
// 构建反应物分子结构
let reactants = build_molecules('A-B-C');
// 设置反应条件
let temperature = 400;
let pressure = 1.0;
// 模拟反应过程
let products = simulate_reaction(reactants, temperature, pressure);
// 输出反应产物结构
console.log(products);
```
通过这个实例,我们可以模拟化学反应的具体过程,探讨反应路径和产物生成情况,有助于理解化学反应的动力学和热力学特性。
通过以上实例,我们可以看到LAMMPS软件在分子动力学模拟中的强大功能,能够帮助科研人员深入研究物质性质和反应过程。
# 6. LAMMPS在科研和工程领域的应用展望
分子动力学模拟软件LAMMPS在科研和工程领域有着广泛的应用前景,下面将讨论几个重要领域中LAMMPS的应用情况及未来展望。
#### 6.1 生物医药研究中的应用
在生物医药领域,LAMMPS可以用于模拟蛋白质、药物与受体的相互作用,帮助科学家们了解药物的作用机理、药效改进以及疾病治疗方案的设计。通过分子动力学模拟,研究人员可以观察生物大分子的结构变化,预测药物分子的扩散路径和结合方式,从而加速药物研发过程。未来,随着计算性能的提升和算法的改进,LAMMPS在生物医药领域的应用将更加深入和广泛。
#### 6.2 新材料设计与开发
在材料科学领域,LAMMPS可以用于模拟材料的力学性能、热学性质、电子结构等方面的特性。科研人员可以通过模拟不同原子结构及相互作用条件下材料的性能,辅助设计新型材料、预测材料的稳定性和响应行为。未来,LAMMPS在新材料设计与开发中的应用将有助于加速材料创新速度,推动材料科学领域的发展。
#### 6.3 环境与能源领域的应用前景
在环境与能源领域,LAMMPS可用于模拟纳米材料在环境中的传输行为、催化剂的反应机理、能源材料的性能等方面。通过模拟,研究人员可以更好地理解材料在复杂环境下的性能,优化环境治理技术和能源材料的设计,为可持续发展提供技术支持。未来,LAMMPS在环境与能源领域的应用前景将推动理论研究与实际应用相结合,促进环保和能源可持续利用领域的进步。

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏"修复墙"旨在探讨如何利用LAMMPS这款强大的分子动力学模拟软件来构建、分析和优化不同类型的墙体模拟系统。从介绍LAMMPS的基础知识,解析粒子间力的作用机制,到探索墙体的应用场景和动力学特性,以及分析墙体与粒子之间的复杂相互作用,本专栏涵盖了从软墙到硬墙、从碰撞事件处理到温度控制的各个方面。读者将了解如何使用fix命令实现柔软墙的模拟,解析不同势函数对墙体相互作用的影响,以及探索墙体在模拟中的稳定性和热传导性能。通过介绍LAMMPS组件manifold,读者还能深入理解墙的多孔结构模拟以及反射边界对模拟结果的影响。本专栏旨在帮助读者全面掌握LAMMPS在墙体模拟中的应用技术和方法,为相关领域的研究和工程实践提供有力支持。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【实时系统空间效率】:确保即时响应的内存管理技巧

# 1. 实时系统的内存管理概念
在现代的计算技术中,实时系统凭借其对时间敏感性的要求和对确定性的追求,成为了不可或缺的一部分。实时系统在各个领域中发挥着巨大作用,比如航空航天、医疗设备、工业自动化等。实时系统要求事件的处理能够在确定的时间内完成,这就对系统的设计、实现和资源管理提出了独特的挑战,其中最为核心的是内存管理。
内存管理是操作系统的一个基本组成部
【算法竞赛中的复杂度控制】:在有限时间内求解的秘籍
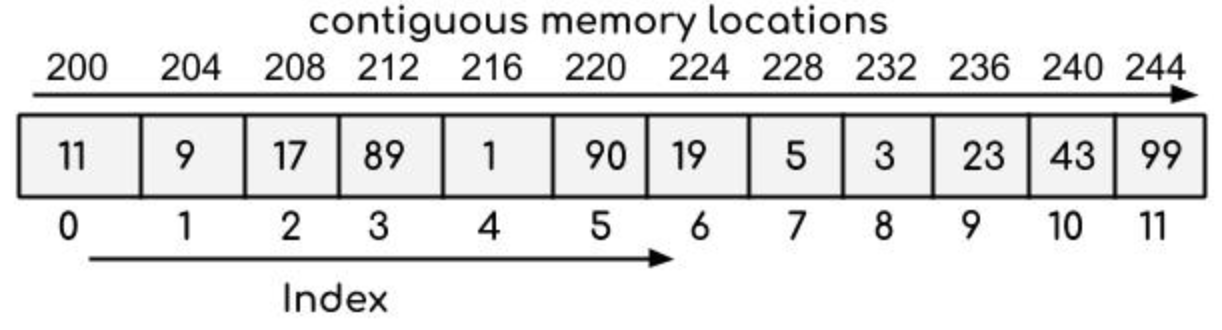
# 1. 算法竞赛中的时间与空间复杂度基础
## 1.1 理解算法的性能指标
在算法竞赛中,时间复杂度和空间复杂度是衡量算法性能的两个基本指标。时间复杂度描述了算法运行时间随输入规模增长的趋势,而空间复杂度则反映了算法执行过程中所需的存储空间大小。理解这两个概念对优化算法性能至关重要。
## 1.2 大O表示法的含义与应用
大O表示法是用于描述算法时间复杂度的一种方式。它关注的是算法运行时
机器学习性能评估:时间复杂度在模型训练与预测中的重要性
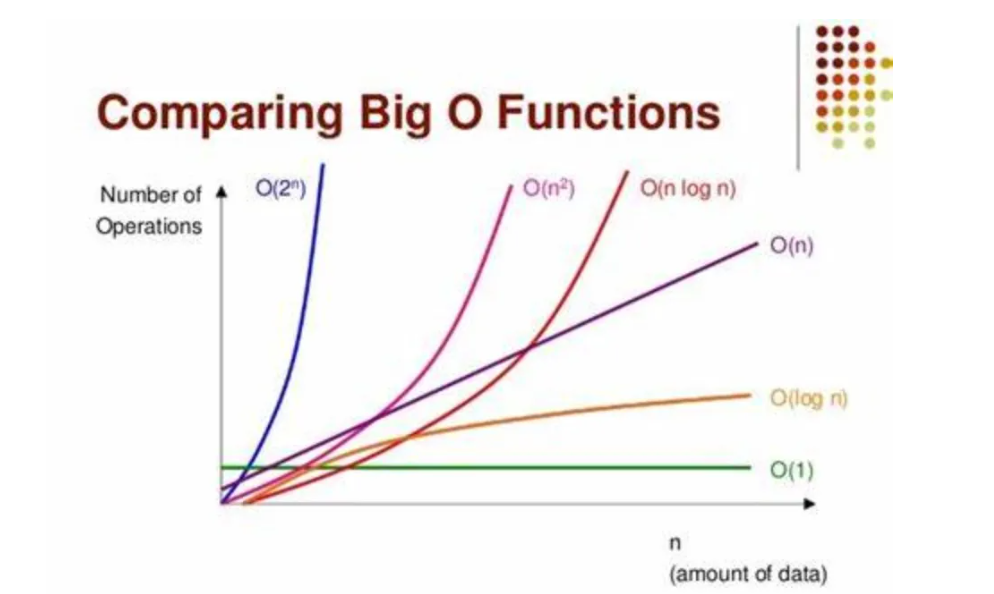
# 1. 机器学习性能评估概述
## 1.1 机器学习的性能评估重要性
机器学习的性能评估是验证模型效果的关键步骤。它不仅帮助我们了解模型在未知数据上的表现,而且对于模型的优化和改进也至关重要。准确的评估可以确保模型的泛化能力,避免过拟合或欠拟合的问题。
## 1.2 性能评估指标的选择
选择正确的性能评估指标对于不同类型的机器学习任务至关重要。例如,在分类任务中常用的指标有
激活函数理论与实践:从入门到高阶应用的全面教程

# 1. 激活函数的基本概念
在神经网络中,激活函数扮演了至关重要的角色,它们是赋予网络学习能力的关键元素。本章将介绍激活函数的基础知识,为后续章节中对具体激活函数的探讨和应用打下坚实的基础。
## 1.1 激活函数的定义
激活函数是神经网络中用于决定神经元是否被激活的数学函数。通过激活函数,神经网络可以捕捉到输入数据的非线性特征。在多层网络结构
时间序列分析的置信度应用:预测未来的秘密武器
# 1. 时间序列分析的理论基础
在数据科学和统计学中,时间序列分析是研究按照时间顺序排列的数据点集合的过程。通过对时间序列数据的分析,我们可以提取出有价值的信息,揭示数据随时间变化的规律,从而为预测未来趋势和做出决策提供依据。
## 时间序列的定义
时间序列(Time Series)是一个按照时间顺序排列的观测值序列。这些观测值通常是一个变量在连续时间点的测量结果,可以是每秒的温度记录,每日的股票价
【损失函数与随机梯度下降】:探索学习率对损失函数的影响,实现高效模型训练

# 1. 损失函数与随机梯度下降基础
在机器学习中,损失函数和随机梯度下降(SGD)是核心概念,它们共同决定着模型的训练过程和效果。本
学习率对RNN训练的特殊考虑:循环网络的优化策略

# 1. 循环神经网络(RNN)基础
## 循环神经网络简介
循环神经网络(RNN)是深度学习领域中处理序列数据的模型之一。由于其内部循环结
Epochs调优的自动化方法
# 1. Epochs在机器学习中的重要性
机器学习是一门通过算法来让计算机系统从数据中学习并进行预测和决策的科学。在这一过程中,模型训练是核心步骤之一,而Epochs(迭代周期)是决定模型训练效率和效果的关键参数。理解Epochs的重要性,对于开发高效、准确的机器学习模型至关重要。
在后续章节中,我们将深入探讨Epochs的概念、如何选择合适值以及影响调优的因素,以及如何通过自动化方法和工具来优化Epochs的设置,从而
【批量大小与存储引擎】:不同数据库引擎下的优化考量

# 1. 数据库批量操作的理论基础
数据库是现代信息系统的核心组件,而批量操作作为提升数据库性能的重要手段,对于IT专业人员来说是不可或缺的技能。理解批量操作的理论基础,有助于我们更好地掌握其实践应用,并优化性能。
## 1.1 批量操作的定义和重要性
批量操作是指在数据库管理中,一次性执行多个数据操作命
极端事件预测:如何构建有效的预测区间
# 1. 极端事件预测概述
极端事件预测是风险管理、城市规划、保险业、金融市场等领域不可或缺的技术。这些事件通常具有突发性和破坏性,例如自然灾害、金融市场崩盘或恐怖袭击等。准确预测这类事件不仅可挽救生命、保护财产,而且对于制定应对策略和减少损失至关重要。因此,研究人员和专业人士持
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )