探索单细胞转录组数据中细胞亚群的发现与标记的R语言方法
发布时间: 2024-04-02 04:14:08 阅读量: 70 订阅数: 32 
# 1. Ⅰ. 前言
## A. 单细胞转录组数据分析的背景与意义
## B. R语言在单细胞转录组数据分析中的作用与优势
# 2. 单细胞转录组数据预处理
A. 数据质控与过滤
B. 数据归一化与批次效应消除
C. 细胞特征筛选与降维处理
# 3. III. 细胞亚群发现与聚类分析
A. 基于降维技术的细胞亚群发现
在单细胞转录组数据分析中,常用的降维技术包括主成分分析(PCA)、t分布邻域嵌入(t-SNE)和Uniform Manifold Approximation and Projection(UMAP)等。这些降维技术可以帮助将高维数据转换为更易于可视化和分析的低维空间,从而更好地揭示细胞之间的相似性和差异性。
代码示例(以Python为例):
```python
from sklearn.decomposition import PCA
import umap
import seaborn as sns
# 使用PCA进行数据降维
pca = PCA(n_components=50)
data_pca = pca.fit_transform(data)
# 使用UMAP进行数据降维
umap_model = umap.UMAP()
data_umap = umap_model.fit_transform(data)
# 展示降维后的数据分布
sns.scatterplot(x=data_umap[:, 0], y=data_umap[:, 1], hue=labels)
```
B. 聚类算法在细胞亚群识别中的应用
聚类是一种常用的无监督学习方法,用于将数据集中相似的样本划分到同一类别中。在单细胞转录组数据分析中,聚类算法能够帮助识别细胞亚群并进行进一步的分析。常用的聚类算法包括K均值聚类、层次聚类和DBSCAN等。
代码示例(以Python为例):
```python
from sklearn.cluster import KMeans
import hdbscan
# 使用K均值聚类进行细胞亚群识别
kmeans = KMeans(n_clusters=3, random_state=0)
cluster_labels = kmeans.fit_predict(data)
# 使用HDBSCAN进行细胞亚群识别
clusterer = hdbscan.HDBSCAN(min_cluster_size=50)
cluster_labels = clusterer.fit_predict(data)
```
C. 可视化展示细胞亚群的分布情况
在细胞亚群分析的过程中,通过可视化工具将不同细胞亚群的分布情况展现出来,有助于直观地理解数据和结果。常用的可视化方式包括散点图、热图、曲线图等。
代码示例(以Python为例):
```python
import matplotlib.pyplot as plt
# 绘制细胞亚群的散点图
plt.figure(figsize=(8,
```

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏深入探讨了R单细胞转录组分析的各个方面,从介绍R语言在单细胞转录组中的基础应用到探讨数据质控、降维分析、细胞聚类、差异表达基因分析等多个环节,全面展现了R语言在单细胞转录组领域的重要性和应用广泛性。文章涵盖了实验流程概述、数据预处理、数据解读、功能富集分析、细胞亚群发现等诸多内容,并展示了丰富的实际案例和操作示范。无论是对于初学者还是有经验的研究者,本专栏都提供了相当丰富和实用的指导,助力他们更好地应用R语言进行单细胞转录组数据的分析与挖掘,为深入理解细胞的功能、发育轨迹以及调控机制提供了强有力的工具支持。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【OBDD技术深度剖析】:硬件验证与软件优化的秘密武器

# 摘要
有序二元决策图(OBDD)是一种广泛应用于硬件验证、软件优化和自动化测试的高效数据结构。本文首先对OBDD技术进行了概述,并深入探讨了其理论基础,包括基本概念、数学模型、结构分析和算法复杂性。随后,本文重点讨论了OBDD在硬件验证与软件优化领域的具体应用,如规范表示、功能覆盖率计算、故障模拟、逻辑分析转换、程序验证和测试用例生成。最后,文章分析了OBDD算法在现代
【微服务架构的挑战与对策】:从理论到实践
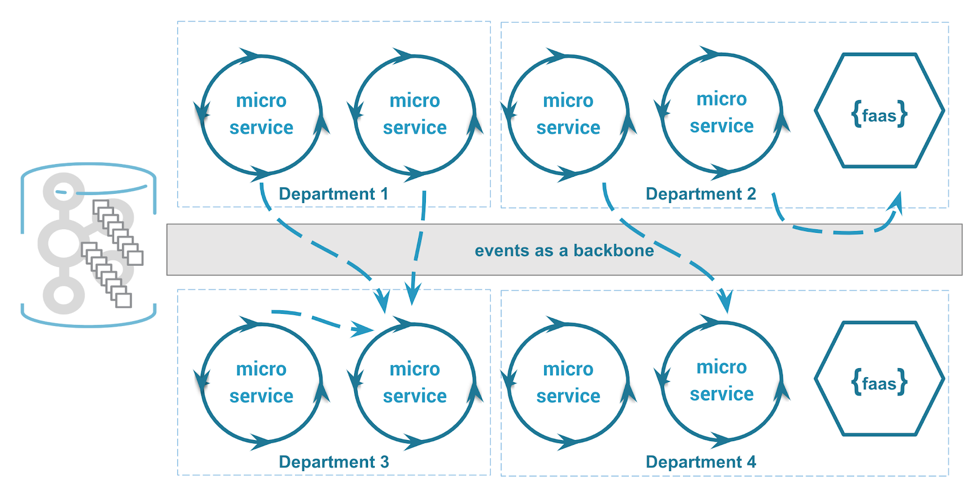
# 摘要
微服务架构作为一种现代化的软件架构方式,通过服务的划分和分布式部署,提高了应用的灵活性和可扩展性。本文从基本概念和原则出发,详细探讨了微服务架构的技术栈和设计模式,包括服务注册与发现、负载均衡、通信机制以及设计模式。同时,文章深入分析了实践中的挑战,如数据一致性、服务治理、安全问题等。在优化策略方面,本文讨论了性能、可靠性和成本控制的改进方法。最后,文章展望了微服务架构的未来趋势,包括服
RadiAnt DICOM Viewer错误不再难:专家解析常见问题与终极解决方案
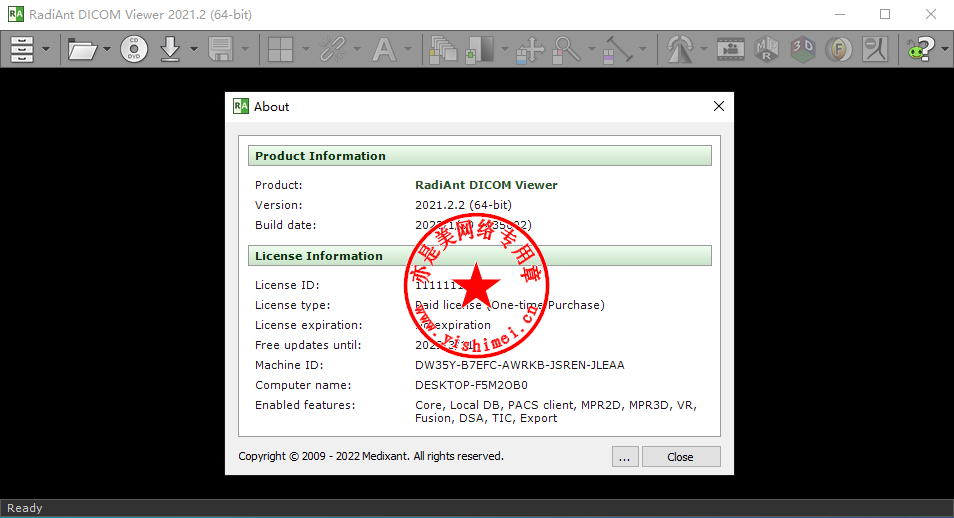
# 摘要
本文对RadiAnt DICOM Viewer这款专业医学影像软件进行了全面的介绍与分析。首先概述了软件的基本功能和常见使用问题,接着深入探讨了软件的错误分析和解决策略,包括错误日志的分析方法、常见错误原因以及理论上的解决方案。第四章提供了具体的终极解决方案实践,包括常规问题和高级问题的解决步骤、预防措施与最佳实践。最后,文章展望了软件未来的优化建议和用户交互提升策略,并预测了技术革新和行业应
macOS用户必看:JDK 11安装与配置的终极指南

# 摘要
本文全面介绍了JDK 11的安装、配置、高级特性和性能调优。首先概述了JDK 11的必要性及其新特性,强调了其在跨平台安装和环境变量配置方面的重要性。随后,文章深入探讨了配置IDE和使用JShell进行交互式编程的实践技巧,以及利用Maven和Gradle构建Java项目的具体方法。在高级特性部分,本文详细介绍了新HTTP Client API的使用、新一代垃圾收集器的应用,以及
华为产品开发流程揭秘:如何像华为一样质量与效率兼得
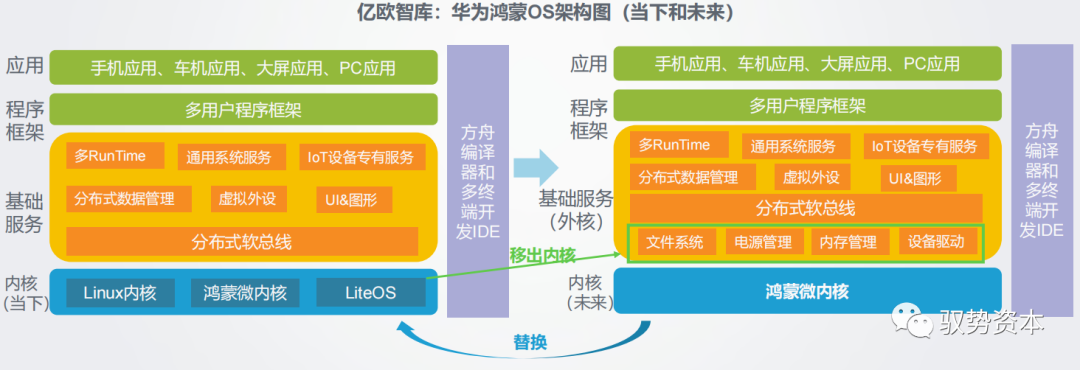
# 摘要
本文详细探讨了华为公司产品开发流程的理论与实践,包括产品生命周期管理理论、集成产品开发(IPD)理论及高效研发组织结构理论的应用。通过对华为市场需求分析、产品规划、项目管理、团队协作以及质量控制和效率优化等关键环节的深入分析,揭示了华为如何通过其独特的开发流程实现产品创新和市场竞争力的提升。本文还着重评估了华为产品的
无线通信深度指南:从入门到精通,揭秘信号衰落与频谱效率提升(权威实战解析)
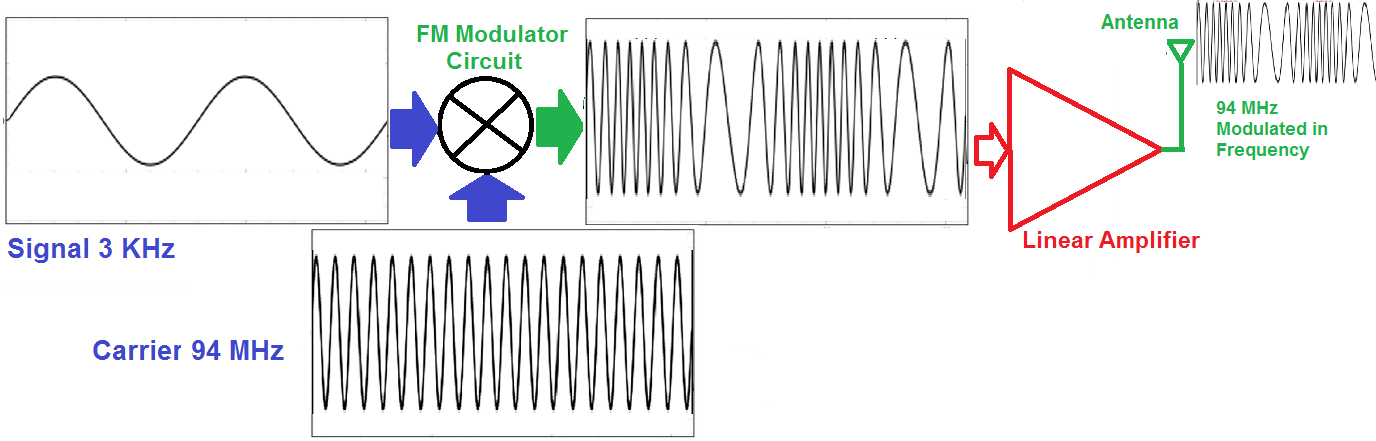
# 摘要
本文深入探讨了无线通信中的频谱效率和信号衰落问题,从基础理论到实用技术进行了全面分析。第一章介绍了无线通信基础及信号衰落现象,阐述了无线信号的传播机制及其对通信质量的影响。第二章聚焦于频谱效率提升的理论基础,探讨了提高频谱效率的策略与方法。第三章则详细讨论了信号调制与解调技
【HOMER最佳实践分享】:行业领袖经验谈,提升设计项目的成功率

# 摘要
本文全面介绍了HOMER项目管理的核心概念、理论基础、实践原则、设计规划技巧、执行监控方法以及项目收尾与评估流程。首先概述了HOMER项目的管理概述,并详细阐释了其理论基础,包括生命周期模型和框架核心理念。实践原则部分强调了明确目标、资源优化和沟通的重要性。设计与规划技巧章节则深入探讨了需求分析、设计方案的迭代、风险评估与应对策略。执行与监控部分着重于执行计划、团队协作、进度跟踪、成本控制和问题解决。最后,在项目收尾与评估章节中,本文涵盖了交付流
【SCSI Primary Commands的终极指南】:SPC-5基础与核心概念深度解析
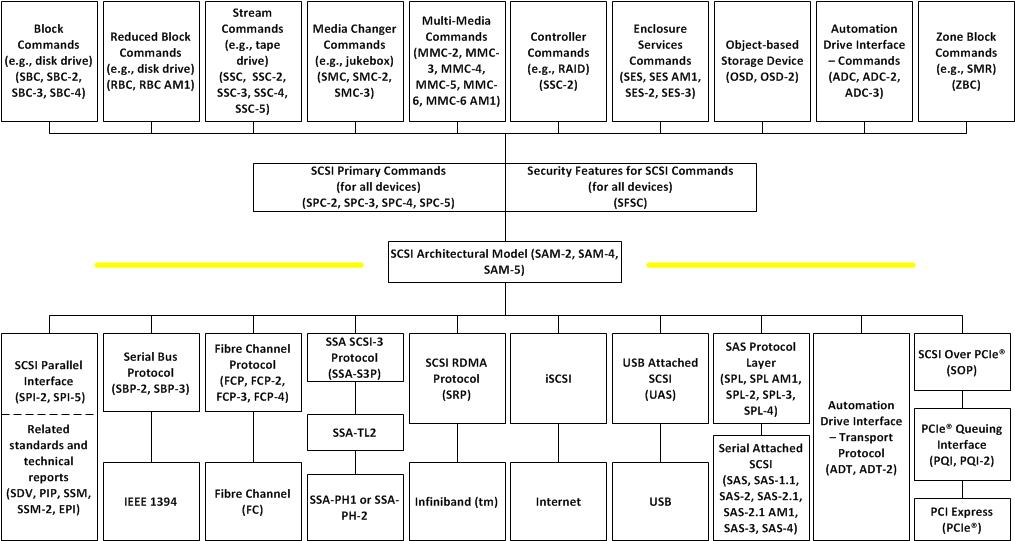
# 摘要
本文系统地探讨了SCSI协议与SPC标准的发展历程、核心概念、架构解析以及在现代IT环境中的应用。文章详细阐述了SPC-5的基本概念、命令模型和传输协议,并分析了不同存储设备的特性、LUN和目标管理,以及数据保护与恢复的策略。此外,本文还讨论了SPC-5在虚拟化环境、云存储中的实施及其监控与诊断工具,展望了SPC-5的技术趋势、标准化扩展和安全性挑战,为存储协议的发展和应用提供了深入的见解。
# 关键字
SCSI协议;S
【工业自动化新星】:CanFestival3在自动化领域的革命性应用

# 摘要
CanFestival3作为一款流行的开源CANopen协议栈,在工业自动化领域扮演着关键角色。本文首先概述了CanFestival3及其在工业自动化中的重要性,随后深入分析其核心原理与架构,包括协议栈基础、配置与初始化以及通信机制。文章详细介绍了CanFestival3在不同工业应用场景中的实践应用案例,如制造业和智慧城市,强调了其对机器人控制系统
【海康威视VisionMaster SDK秘籍】:构建智能视频分析系统的10大实践指南

# 摘要
本文详细介绍了海康威视VisionMaster SDK的核心概念、基础理论以及实际操作指南,旨在为开发者提供全面的技术支持和应用指导。文章首先概述了智能视频分析系统的基础理论和SDK架构,紧接着深入探讨了实际操作过程中的环境搭建、核心功能编程实践和系统调试。此外,本文还分享了智能视频分析系统的高级应用技巧,如多通道视频同步分析、异常行为智能监测和数据融合
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )