R语言在单细胞转录组数据解读中的数据挖掘技术
发布时间: 2024-04-02 04:17:11 阅读量: 32 订阅数: 26 
# 1. 单细胞转录组数据分析简介
- **1.1 什么是单细胞转录组数据?**
- **1.2 单细胞转录组数据在生物学研究中的应用**
- **1.3 数据挖掘在单细胞转录组数据解读中的重要性**
# 2. R语言在生物信息学中的应用概述
R语言作为一种开源的统计计算和数据可视化工具,在生物信息学领域具有广泛的应用。其强大的数据处理能力和丰富的生物信息学相关包使其成为研究人员喜爱的工具之一。
### 2.1 R语言在生物信息学领域的优势
R语言具有以下优势:
- 开源免费:R语言的开源性使得科研人员可以自由获取和使用。
- 强大的数据分析功能:R语言有丰富的内置函数和包,可以进行数据预处理、统计分析和可视化。
- 庞大的社区支持:R语言拥有庞大的用户社区和活跃的开发者,用户可以快速获取帮助并分享代码和经验。
### 2.2 常用的R包及其功能介绍
在生物信息学领域,有一些常用的R包包括:
- `Seurat`:用于单细胞转录组数据的整合、分析和可视化。
- `DESeq2`:用于差异表达基因分析。
- `clusterProfiler`:用于功能富集分析。
这些R包提供了丰富的功能,帮助研究人员在生物信息学研究中高效地处理和分析数据。
### 2.3 如何在R环境中进行单细胞转录组数据分析
在R环境中进行单细胞转录组数据分析通常包括以下步骤:
1. 数据导入:使用`readRDS()`或其他函数导入单细胞转录组数据。
2. 数据预处理:包括数据质控、归一化、批次效应纠正等。
3. 基因表达模式分析:进行基因差异表达分析、聚类分析等。
4. 细胞类型识别和功能分析:识别细胞类型、进行功能富集分析等。
5. 结果可视化:使用`ggplot2`等包进行数据可视化展示。
通过以上步骤,研究人员可以全面地分析单细胞转录组数据,挖掘其中隐藏的生物学信息。R语言在这一过程中发挥着重要的作用,为生物信息学研究提供了强大的工具支持。
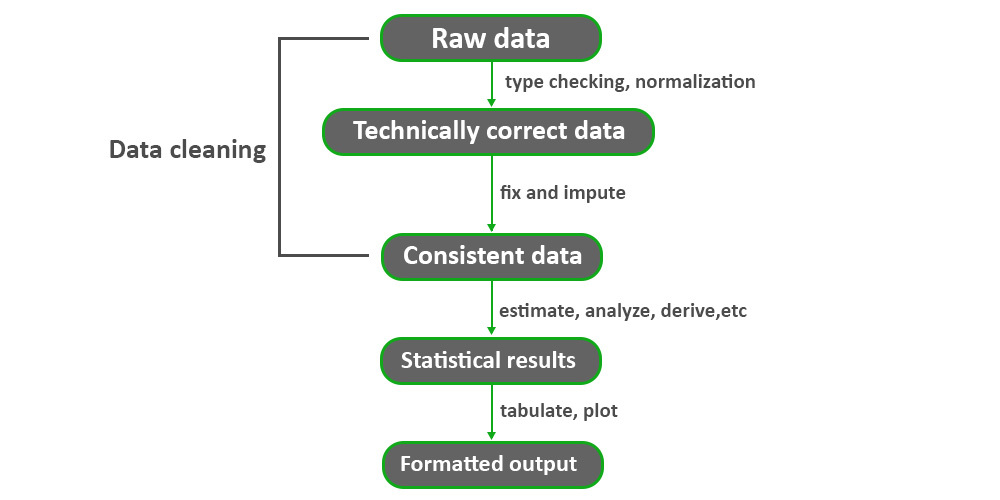
# 3. 单细胞数据预处理
在单细胞转录组数据分析中,数据预处理是非常关键的一步。本章将介绍在R语言环境中进行单细胞数据预处理的具体步骤和技术。
#### 3.1 数据质控和过滤
在进行单细胞数据分析之前,首先需要进行数据的质控和过滤。这一步旨在排除可能存在的噪音数据和低质量细胞,确保后续分析的准确性和可靠性。常见的数据质控和过滤方法包括:
```R
# 导入单细胞数据
sc_data <- readRDS("sc_data.rds")
# 数据质控
filtered_data <- scater::filterCells(sc_data, subset.row = scater::rowData(sc_data)$nFeature_RNA > 200 & scater::rowSums(scater::assay(sc_data) > 0) > 500)
# 低质量细胞过滤
filtered_data <- scater::filterCells(filtered_data, subset.row = scater::rowData(filtered_data)$percent.mt < 20)
```
#### 3.2 数据归一化和批次效应的纠正
数据归一化是为了消除不同细胞之间的技术差异,使得数据更具有可比性。同时,批次效应的纠正可以消除实验中可能存在的批次效应,保证数据的一致性。在R语言中,可以使用以下代码进行数据的归一化和批次效应的纠正:
```R
# 数据归一化
normalized_data <- scran::computeSumFactors(filtered_data)
# 批次效应纠正
corrected_data <- sva::ComBat(dat = scater::assay(normalized_data), batch = scater::rowData(normalized_data)$batch)
```
#### 3.3 数据降维和可视化技术
数据降维是为了将高维的数据转换为低维空间,便于后续的数据分析和可视化展示。常用的降维算法包括PCA和t-SNE。在R语言中,可以使用以下代码进行数据降维和可视化:
```R
# 数据降维
dim_reduced_data <- irlba::irlba(scater::assay(
```

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏深入探讨了R单细胞转录组分析的各个方面,从介绍R语言在单细胞转录组中的基础应用到探讨数据质控、降维分析、细胞聚类、差异表达基因分析等多个环节,全面展现了R语言在单细胞转录组领域的重要性和应用广泛性。文章涵盖了实验流程概述、数据预处理、数据解读、功能富集分析、细胞亚群发现等诸多内容,并展示了丰富的实际案例和操作示范。无论是对于初学者还是有经验的研究者,本专栏都提供了相当丰富和实用的指导,助力他们更好地应用R语言进行单细胞转录组数据的分析与挖掘,为深入理解细胞的功能、发育轨迹以及调控机制提供了强有力的工具支持。
专栏目录
最低0.47元/天 解锁专栏
买1年送3个月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【数据清洗艺术】:R语言density函数在数据清洗中的神奇功效

# 1. 数据清洗的必要性与R语言概述
## 数据清洗的必要性
在数据分析和挖掘的过程中,数据清洗是一个不可或缺的环节。原始数据往往包含错误、重复、缺失值等问题,这些问题如果不加以处理,将严重影响分析结果的准确性和可靠性。数据清洗正是为了纠正这些问题,提高数据质量,从而为后续的数据分析和模型构建打下坚实的基础。
## R语言概述
R语言是一种用于统计分析
【保险行业extRemes案例】:极端值理论的商业应用,解读行业运用案例
# 1. 极端值理论概述
极端值理论是统计学的一个重要分支,专注于分析和预测在数据集中出现的极端情况,如自然灾害、金融市场崩溃或保险索赔中的异常高额索赔。这一理论有助于企业和机构理解和量化极端事件带来的风险,并设计出更有效的应对策略。
## 1.1 极端值理论的定义与重要性
极端值理论提供了一组统计工具,
R语言数据分析高级教程:从新手到aov的深入应用指南
# 1. R语言基础知识回顾
## 1.1 R语言简介
R语言是一种开源编程语言和软件环境,特别为统计计算和图形表示而设计。自1997年由Ross Ihaka和Robert Gentleman开发以来,R已经成为数据科学领域广受欢迎的工具。它支持各种统计技术,包括线性与非线性建模、经典统计测试、时间序列分析、分类、聚类等,并且提供了强大的图形能力。
## 1.2 安装与配置R环境
要开始使用R语言,首先需要在计算机上安装R环境。用户可以访问官方网站
【R语言时间序列预测大师】:利用evdbayes包制胜未来
# 1. R语言与时间序列分析基础
在数据分析的广阔天地中,时间序列分析是一个重要的分支,尤其是在经济学、金融学和气象学等领域中占据
【R语言编程实践手册】:evir包解决实际问题的有效策略

# 1. R语言与evir包概述
在现代数据分析领域,R语言作为一种高级统计和图形编程语言,广泛应用于各类数据挖掘和科学计算场景中。本章节旨在为读者提供R语言及其生态中一个专门用于极端值分析的包——evir——的基础知识。我们从R语言的简介开始,逐步深入到evir包的核心功能,并展望它在统计分析中的重要地位和应用潜力。
首先,我们将探讨R语言作为一种开源工具的优势,以及它如何在金融
【R语言统计推断】:ismev包在假设检验中的高级应用技巧
# 1. R语言与统计推断基础
## 1.1 R语言简介
R语言是一种用于统计分析、图形表示和报告的编程语言和软件环境。由于其强大的数据处理能力、灵活的图形系统以及开源性质,R语言被广泛应用于学术研究、数据分析和机器学习等领域。
## 1.2 统计推断基础
统计推断是统计学中根据样本数据推断总体特征的过程。它包括参数估计和假设检验两大主要分支。参数估计涉及对总体参数(如均值、方差等)的点估计或区间估计。而
R语言数据包个性化定制:满足复杂数据分析需求的秘诀

# 1. R语言简介及其在数据分析中的作用
## 1.1 R语言的历史和特点
R语言诞生于1993年,由新西兰奥克兰大学的Ross Ihaka和Robert Gentleman开发,其灵感来自S语言,是一种用于统计分析、图形表示和报告的编程语言和软件环境。R语言的特点是开源、功能强大、灵活多变,它支持各种类型的数据结
【R语言极值事件预测】:评估和预测极端事件的影响,evd包的全面指南

# 1. R语言极值事件预测概览
R语言,作为一门功能强大的统计分析语言,在极值事件预测领域展现出了其独特的魅力。极值事件,即那些在统计学上出现概率极低,但影响巨大的事件,是许多行业风险评估的核心。本章节,我们将对R语言在极值事件预测中的应用进行一个全面的概览。
首先,我们将探究极值事
R语言prop.test应用全解析:从数据处理到统计推断的终极指南
# 1. R语言与统计推断简介
统计推断作为数据分析的核心部分,是帮助我们从数据样本中提取信息,并对总体进行合理假设与结论的数学过程。R语言,作为一个专门用于统计分析、图形表示以及报告生成的编程语言,已经成为了数据科学家的常用工具之一。本章将为读者们简要介绍统计推断的基本概念,并概述其在R语言中的应用。我们将探索如何利用R语言强大的统计功能库进行实验设计、数据分析和推断验证。通过对数据的
【R语言t.test实战演练】:从数据导入到结果解读,全步骤解析

# 1. R语言t.test基础介绍
统计学是数据分析的核心部分,而t检验是其重要组成部分,广泛应用于科学研究和工业质量控制中。在R语言中,t检验不仅易用而且功能强大,可以帮助我们判断两组数据是否存在显著差异,或者某组数据是否显著不同于预设值。本章将为你介绍R语言中t.test函数的基本概念和用法,以便你能快速上手并理解其在实际工作中的应用价值。
## 1.1 R语言t.test函数概述
R语言t.test函数是一个
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3个月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )