使用Quantum ESPRESSO进行基础计算
需积分: 10 176 浏览量
更新于2024-07-18
收藏 1.72MB PDF 举报
"如何使用QE进行简单计算"
量子ESPRESSO(Quantum ESPRESSO)是一个开源软件包,专门用于进行第一性原理的材料建模和计算。它基于密度泛函理论(DFT),广泛应用于凝聚态物理、材料科学以及化学等领域,用于研究原子系统的基态性质、能量优化以及结构动力学等。该软件包提供了计算材料电子结构和性质的工具,包括分子动力学、几何优化、能带结构计算等。
1. 关于量子ESPRESSO(Quantum ESPRESSO)分布:
量子ESPRESSO是www.quantum-espresso.org网站上发布的一个计算物理和材料科学软件套件。这个软件包由P. Giannozzi等研究人员开发,旨在提供一个高效、可扩展的平台,用于模拟各种材料的电子结构和动力学行为。
2. 名称“Quantum ESPRESSO”的含义:
“Quantum ESPRESSO”这个名字代表了其核心功能,即通过高效的算法和并行化处理,实现快速而精确的量子力学计算,类似于在咖啡馆享用快速服务(ESPRESSO)一样,快速获取计算结果。
3. 许可证与组织结构:
量子ESPRESSO遵循GNU General Public License(GPL)进行分发,允许用户自由使用、修改和重新分发代码。软件包的组织结构清晰,包含了多个子程序,如WANNIER90用于生成最大局域化 wannier 函数,Pwcond用于计算电导,WanT用于相干输运,Xspectra用于计算X射线近边吸收光谱,GIPAW则用于计算电子顺磁共振(EPR)和核磁共振(NMR)的化学位移。
4. 主要功能:
- 基态总能量计算:这是材料科学中最基础的计算之一,用于确定系统的最低能量状态,通常涉及自洽场迭代。
- 原子结构优化:通过最小化势能面来找到最稳定的原子排列,这涉及到原子坐标和单元参数的调整。
- 分子动力学:模拟系统随时间的动态行为,例如热运动或化学反应过程。
- 能带结构计算:揭示材料的电子结构,预测其电学和光学性质。
- 最大局域化Wannier函数:用于研究材料的拓扑性质和电子关联效应。
5. 扩展功能与未来发展方向:
除了上述功能,量子ESPRESSO还在不断扩展,即将支持如GW方法(GWW)进行更精确的带结构计算,以及时间依赖的密度泛函理论(TD-DFT)研究光诱导的电子过程。
通过量子ESPRESSO,科学家和工程师能够深入理解材料的微观行为,从而设计新的功能材料和设备。学习和掌握量子ESPRESSO的使用,对于进行材料的模拟研究具有至关重要的作用。
Shobhana Narasimhan, JNCASR
12
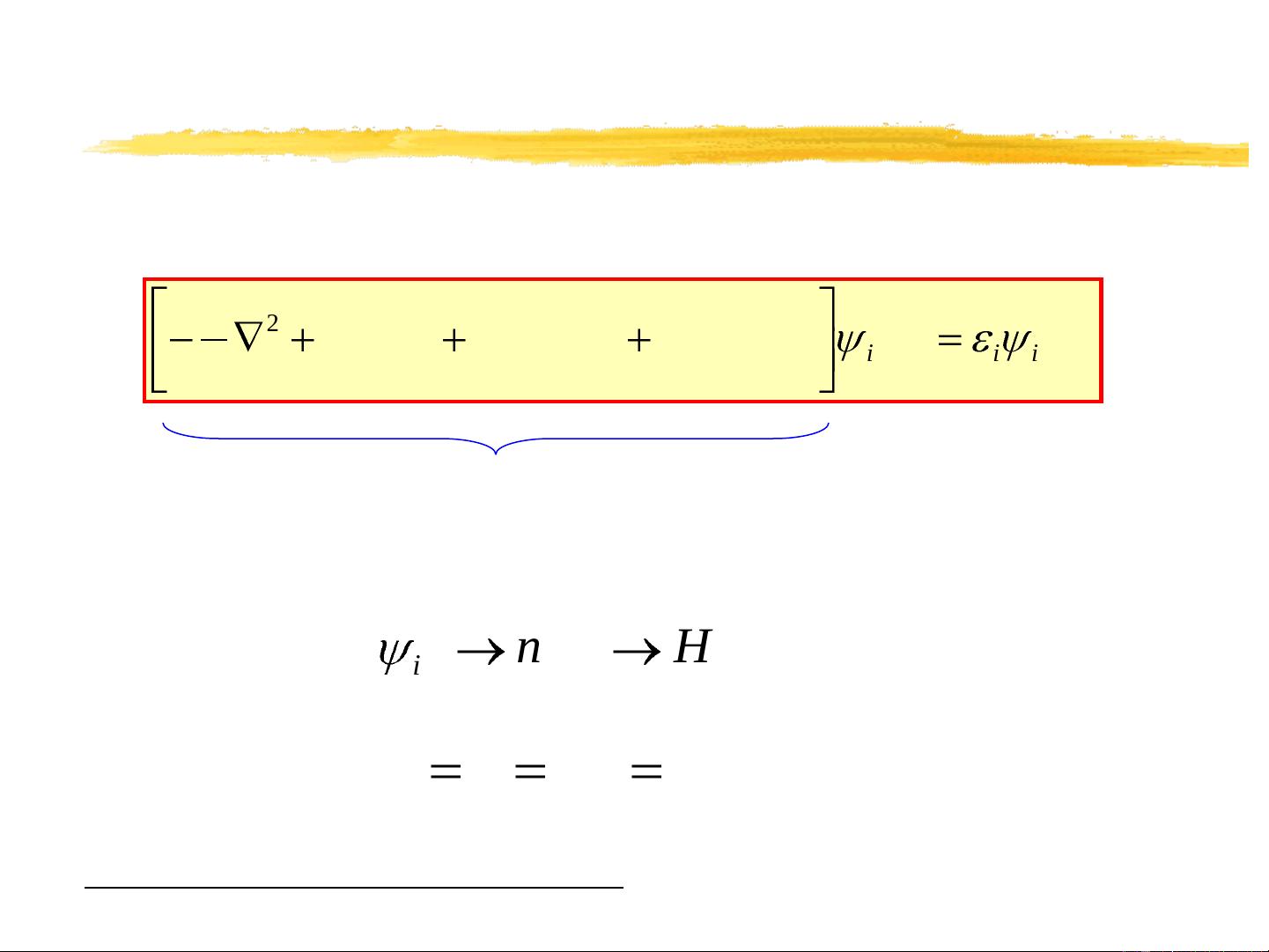
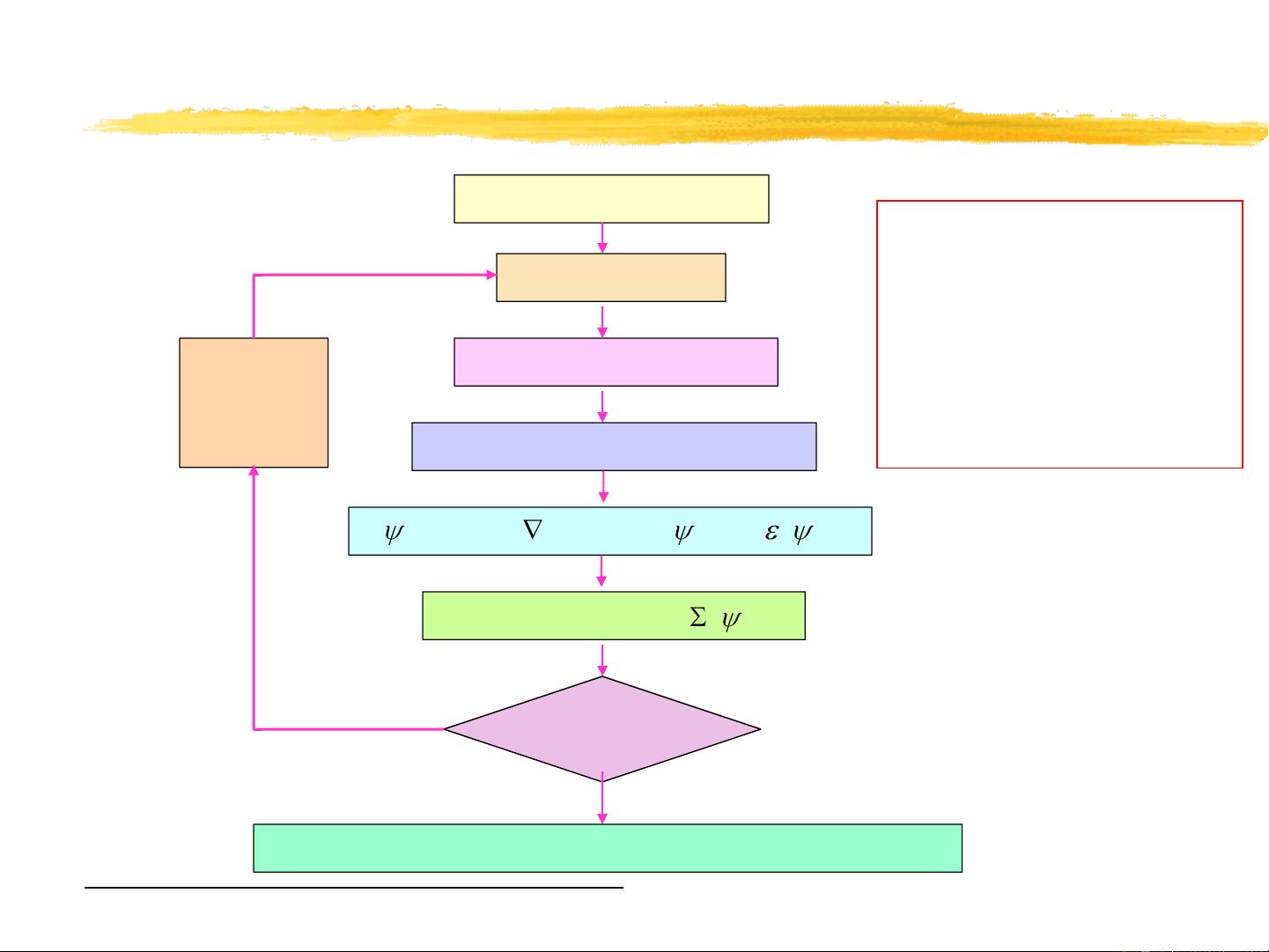
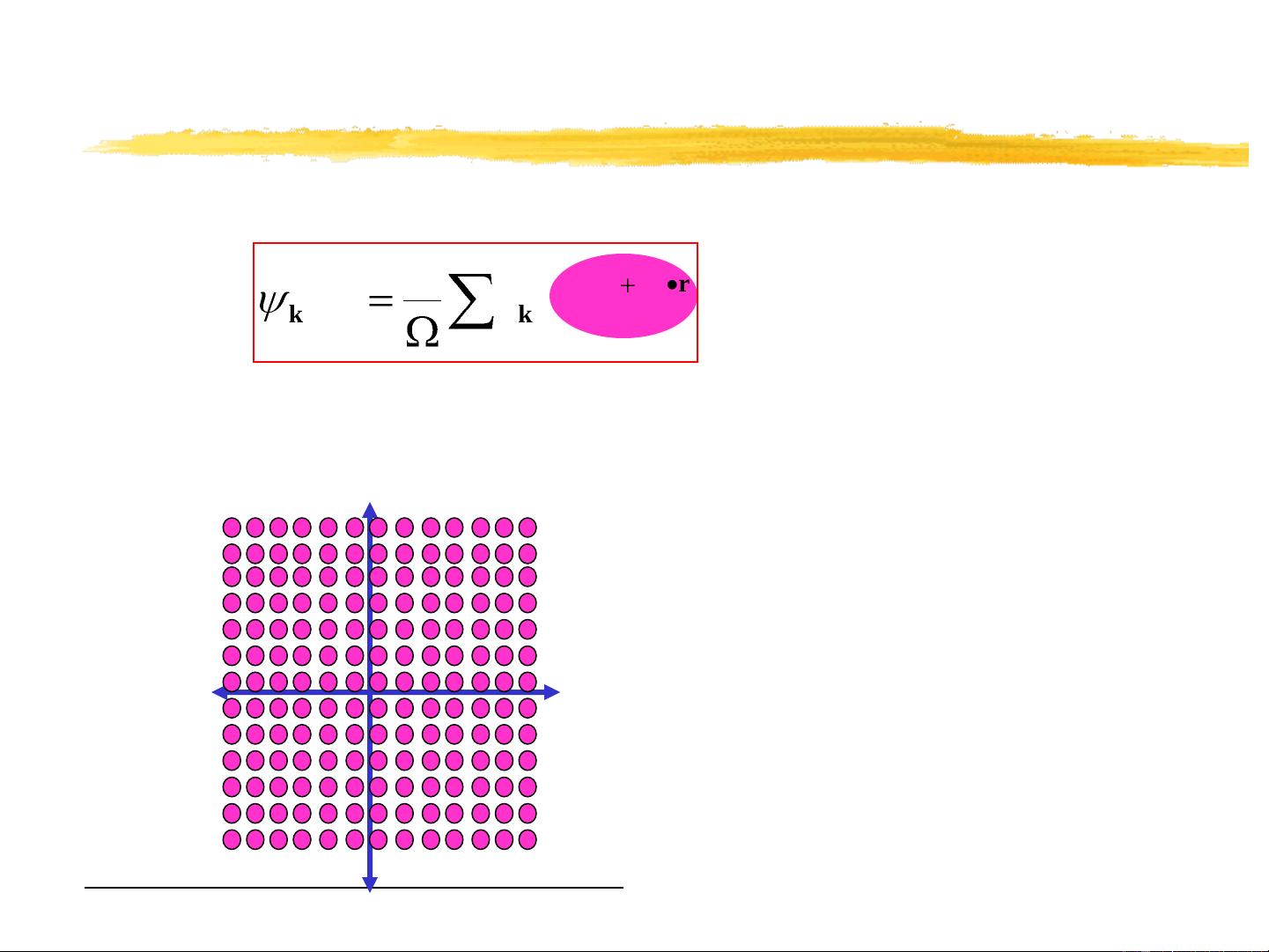
The Kohn-Sham problem
Want to solve the Kohn-Sham equations:
Note that self-consistent solution necessary, as H
depends on solution:
Convention:
)()()]([)]([)(
2
1
2
rrrrr
iiiXCHnuc
nVnVV
H
Hrn
i
)(}{
1
e
me
剩余68页未读,继续阅读
2023-04-04 上传
2023-06-09 上传
2023-05-05 上传
2023-06-09 上传
2023-04-04 上传
2023-06-12 上传
2023-03-30 上传

H^_^Honey
- 粉丝: 1
- 资源: 1
 我的内容管理
展开
我的内容管理
展开







最新资源
- 解决本地连接丢失无法上网的问题
- BIOS报警声音解析:故障原因与解决方法
- 广义均值移动跟踪算法在视频目标跟踪中的应用研究
- C++Builder快捷键大全:高效编程的秘密武器
- 网页制作入门:常用代码详解
- TX2440A开发板网络远程监控系统移植教程:易搭建与通用解决方案
- WebLogic10虚拟内存配置详解与优化技巧
- C#网络编程深度解析:Socket基础与应用
- 掌握Struts1:Java MVC轻量级框架详解
- 20个必备CSS代码段提升Web开发效率
- CSS样式大全:字体、文本、列表样式详解
- Proteus元件库大全:从基础到高级组件
- 74HC08芯片:高速CMOS四输入与门详细资料
- C#获取当前路径的多种方法详解
- 修复MySQL乱码问题:设置字符集为GB2312
- C语言的诞生与演进:从汇编到系统编程的革命
