
为了测试基本的操作流程,我们确定了测试家族数据集。特别的我们集中在核糖开关上,它
具有一定的复杂结构。为了产生一个代表性的RNA家族数据集在我们的研究中用于基因组分
析和结构预测,我们从Rfam数据库中通过以下标准系统地选择了核糖开关家族:
属于一个在Rfam数据库中有超过1000个序列的家族,来提供检测核苷酸协同进化足
够的统计数据
其PDB结构最大有100个核苷酸,使得像Rosetta分子建模工具能够有效的运用
有一个相应的完整PDBX射线衍射结构(每一个包含的残基至少有原子表示),其结
构分辨率小于3埃以便评估我们的方法
如果有多个满足这些条件的PDB结构,我们选择最高精确度的那个。我们发现有六个家族
(见附录表S1)满足这些条件,那些不满足条件的家族则不包含在我们的分析中。有趣的
是,这些核糖开关是很多研究常用的目标[38,41]。
对于这些家族,我们从Rfam数据库中提取了多序列比对和一致的二级结构[42]。为了适用于
特定的RNA序列的结构预测,通过去除关联精选那些一致的二级结构,这在特定序列会不满
足碱基基本配对,并有可能通过添加邻近二级结构的基本配对延伸螺旋链。见附录表S2。
这些选择标准不应被理解为我们的方法的适用性限制。正如附录图SI1所示,尽管使用了至
少250个子样品,DCA提取的关联信息也不会改变多少。整个操作流程同样适用于超过100
个核苷酸的序列,只是在分子建模步骤的计算成本有所增加。
为了证明我们的操作流程除了测试集外仍具有广泛的适用性,我们还分析了两个未知3D结构
的RNA家族,即glnA核糖开关家族RF01739和C4反义RNA家族RF01695。我们提供了未知
结构预测的十个最高的打分聚类,见附录数据(pdb文件),可以进行实验上的检测。
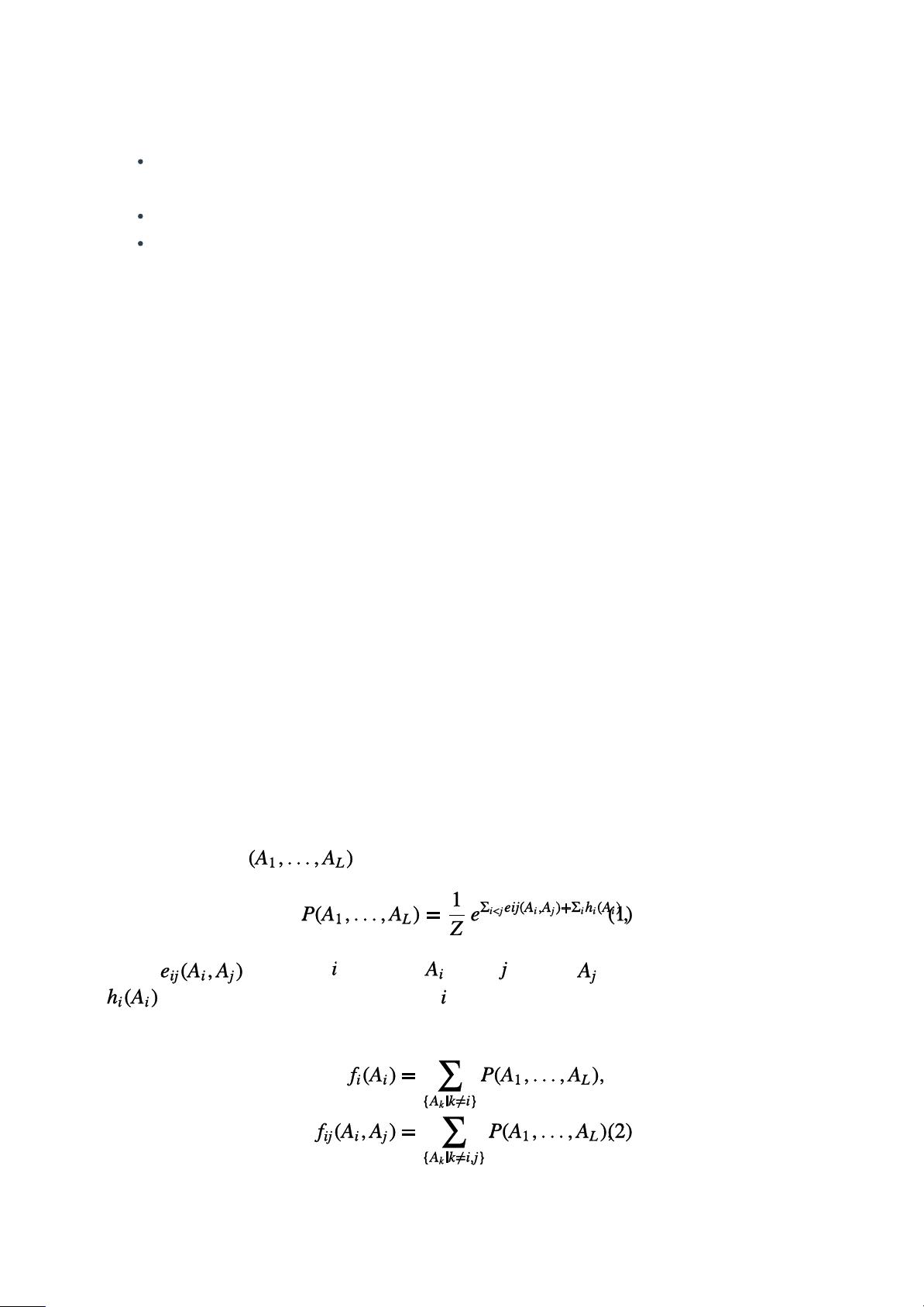
直接耦合分析
接下来我们简单的回顾DCA的主要部分(更多详细的描述包括具体技术细节请见附录,其中
主要步骤[19]是总结和适用于RNA的特异性):DCA旨在通过广义Potts模型(或者说马尔可
夫随机场),MSA作为输入,来模拟序列的变异性。在L个核苷酸或间隔中,它分配一个概
率给每个比对序列 ,
其中, 指的是位于 处的核苷酸 和位于 处核苷酸 之间的直接耦合,并且
是局部偏差(场),仅涉及处于位置 的单个核苷酸。分母Z起到归一化作用。参数必
须调整以便再现从MSA获取的核苷酸实验统计数据:
剩余17页未读,继续阅读

俞林鑫
- 粉丝: 19
- 资源: 288
 我的内容管理
展开
我的内容管理
展开







最新资源
- C++标准程序库:权威指南
- Java解惑:奇数判断误区与改进方法
- C++编程必读:20种设计模式详解与实战
- LM3S8962微控制器数据手册
- 51单片机C语言实战教程:从入门到精通
- Spring3.0权威指南:JavaEE6实战
- Win32多线程程序设计详解
- Lucene2.9.1开发全攻略:从环境配置到索引创建
- 内存虚拟硬盘技术:提升电脑速度的秘密武器
- Java操作数据库:保存与显示图片到数据库及页面
- ISO14001:2004环境管理体系要求详解
- ShopExV4.8二次开发详解
- 企业形象与产品推广一站式网站建设技术方案揭秘
- Shopex二次开发:触发器与控制器重定向技术详解
- FPGA开发实战指南:创新设计与进阶技巧
- ShopExV4.8二次开发入门:解决升级问题与功能扩展
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


