

What's new in Monocle 3
Monocle 3 has been re-engineered to analyze large, complex single-cell datasets. The algorithms at the core of Monocle 3 are highly scalable and
can handle millions of cells. Monocle 3 will add some powerful new features that enable the analysis of organism- or embryo-scale experiments:
A better structured workow to learn developmental trajectories.
Support for the UMAP (https://github.com/lmcinnes/umap) algorithm to initialize trajectory inference.
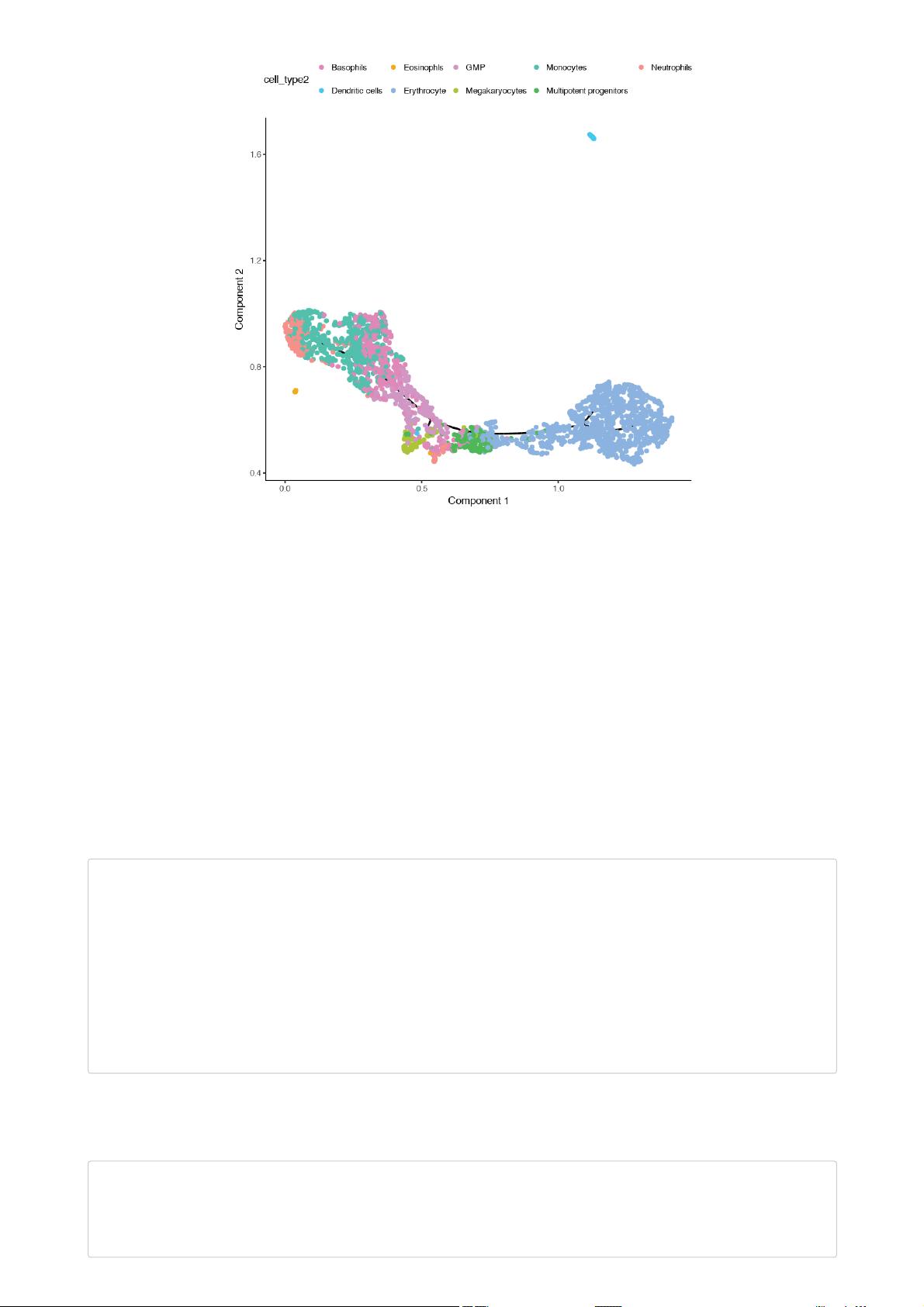
Support for trajectories with multiple roots.
Ways to learn trajectories that have loops or points of convergence.
Algorithms that automatically partition cells to learn disjoint or parallel trajectories using ideas from "approximate graph abstraction"
(https://www.biorxiv.org/content/early/2017/10/25/208819).
A new statistical test for genes that have trajectory-dependent expression.
A 3D interface to visualize trajectories and gene expression.
Monocle 3 is still under active development. This rst early alpha release only includes some of the features listed above. We plan to release
updates to Monocle 3 every few weeks that add the functionality you see mentioned here. Moreover, the above is only a partial list of the new
features being added to Monocle 3, and more may be announced over the next few weeks and months. Note that some of Monocle 2's functions
haven't yet been forward ported to Monocle 3. We welcome users' bug reports, comments and feedback.
The features discussed here will be described in more detail in upcoming manuscripts.
Below, you can nd more details about some of the major new ideas and features in Monocle 3.
Uniform Manifold Approximation and Projection
Recently, Leland McInnes and John Healy introduced UMAP, a powerful new manifold learning technique that we've found works extremely well on
single-cell RNA-seq data. Monocle 3 uses UMAP to embed cells in a low dimensional space, and then uses our principal graph embedding algorithms
to learn a trajectory that ts the cells' UMAP coordinates. This approach enables Monocle 3 to learn very complex, potentially disjoint trajectories
without much parameter optimization. Monocle 3 currently relies on the python implementation of UMAP (https://github.com/lmcinnes/umap). You
can check out the preprint about UMAP here:
McInnes, L, Healy, J, UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction (https://arxiv.org/abs/1802.03426), ArXiv e-
prints 1802.03426, 2018).
In Monocle 3 we provide a standard alone function UMAP to run umap. UMAP is also seamlessly integrated into the reduceDimension function.
Installing Monocle 3
Required software
Monocle runs in the R statistical computing environment (http://www.r-project.org/). You will need R version 3.5 or higher, Bioconductor version 3.7,
and monocle 2.99.0 or higher to have access to the latest features. To install Bioconductor (http://bioconductor.org/install/):
source("http://bioconductor.org/biocLite.R")
biocLite()
Once you've installed Bioconductor, make sure you've installed Monocle 2 and all of its required dependencies:
biocLite("monocle")
Install DDRTree (simple-ppt-like branch) from our GitHub repo:
::install_github("cole-trapnell-lab/DDRTree", ref="simple-ppt-like")
Install the latest version of L1-graph from our GitHub repo:
::install_github("cole-trapnell-lab/L1-graph")
Install several python packages Monocle 3 depends on:
install.packages("reticulate")
library(reticulate)
py_install('umap-learn', pip = T, pip_ignore_installed = T) # Ensure the latest version of UMAP is installed
py_install("louvain")
Then pull the monocle3_alpha branch of the Monocle GitHub repo:
::install_github("cole-trapnell-lab/monocle-release", ref="monocle3_alpha")
Testing the installation
devtools
devtools
devtools



剩余100页未读,继续阅读

SmartCells
- 粉丝: 5
- 资源: 8
 我的内容管理
收起
我的内容管理
收起
- 我的资源
快来上传第一个资源
 我的收益 登录查看自己的收益
我的收益 登录查看自己的收益 我的积分
登录查看自己的积分
我的积分
登录查看自己的积分
 我的C币
登录后查看C币余额
我的C币
登录后查看C币余额
 我的收藏
我的收藏  我的下载
我的下载  下载帮助
下载帮助

会员权益专享




最新资源
- zigbee-cluster-library-specification
- JSBSim Reference Manual
- c++校园超市商品信息管理系统课程设计说明书(含源代码) (2).pdf
- 建筑供配电系统相关课件.pptx
- 企业管理规章制度及管理模式.doc
- vb打开摄像头.doc
- 云计算-可信计算中认证协议改进方案.pdf
- [详细完整版]单片机编程4.ppt
- c语言常用算法.pdf
- c++经典程序代码大全.pdf
- 单片机数字时钟资料.doc
- 11项目管理前沿1.0.pptx
- 基于ssm的“魅力”繁峙宣传网站的设计与实现论文.doc
- 智慧交通综合解决方案.pptx
- 建筑防潮设计-PowerPointPresentati.pptx
- SPC统计过程控制程序.pptx
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈



评论1