

原创】GATK 使用方法详解(包含 bwa 使用)第一部分
(2014-03-03 11:07:29)
转 载 ▼
标签:
gatk
bwa
snp
indel
分类:生物信息
由于新浪博客规定,每篇文章不可超过 2 万字符,因此分 4 篇发布。
一、使用 GATK 前须知事项:
()对 的测试主要使用的是人类全基因组和外显子组的测序数据,而且全部是
基于 数据格式,目前还没有提供其他格式文件(如 )或者实验
设计()的分析方法。
() 是一个应用于前沿科学研究的软件,不断在更新和修正,因此,在使用
进行变异检测时,最好是下载最新的版本,目前的版本是 (
)。下载网站: !!!"#$% %& #!#。
(')在 使用过程中(见下面图),有些步骤需要用到已知变异信息,对于这些
已知变异, 只提供了人类的已知变异信息,可以在 的 () 站点下载
($*"#)。如果要研究的不是人类基因组,需要自行构建已知变
异, 提供了详细的构建方法。
() 在进行 +, 和 -, 的过程中会使用到 软件绘制一些图,因此,在运
行 之前最好先检查一下是否正确安装了 和所需要的包,所需要的包大概
包括
%%、%$、"$、*$、*$*、%#、%$"、$、.+
! 等。如果画图时出现错误,会提示需要安装的包的名称。
/
二、GATK 的使用流程
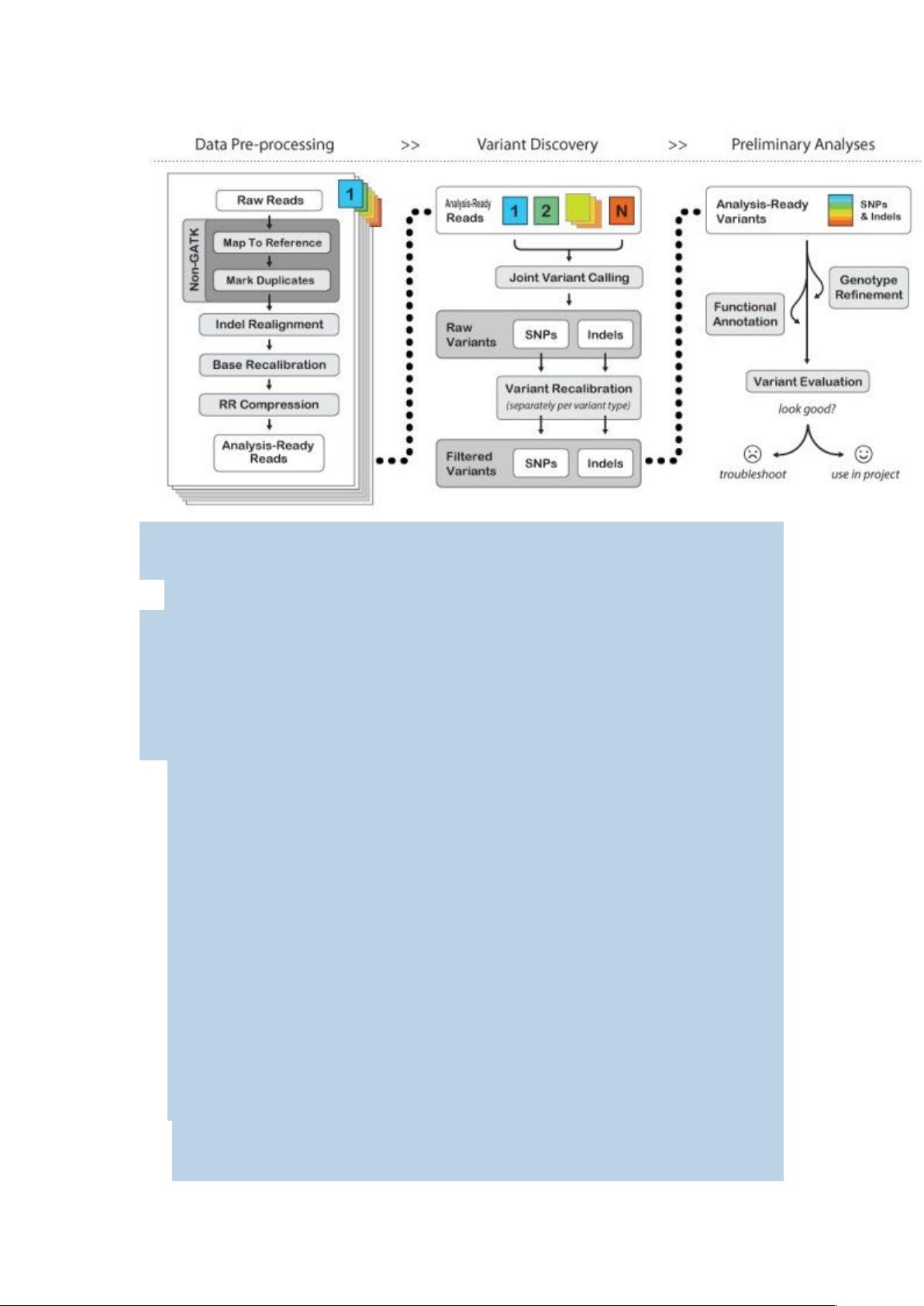

剩余21页未读,继续阅读

rogerzhanglijie
- 粉丝: 305
- 资源: 2
 我的内容管理
收起
我的内容管理
收起
- 我的资源
快来上传第一个资源
 我的收益 登录查看自己的收益
我的收益 登录查看自己的收益 我的积分
登录查看自己的积分
我的积分
登录查看自己的积分
 我的C币
登录后查看C币余额
我的C币
登录后查看C币余额
 我的收藏
我的收藏  我的下载
我的下载  下载帮助
下载帮助

会员权益专享




最新资源
- zigbee-cluster-library-specification
- JSBSim Reference Manual
- c++校园超市商品信息管理系统课程设计说明书(含源代码) (2).pdf
- 建筑供配电系统相关课件.pptx
- 企业管理规章制度及管理模式.doc
- vb打开摄像头.doc
- 云计算-可信计算中认证协议改进方案.pdf
- [详细完整版]单片机编程4.ppt
- c语言常用算法.pdf
- c++经典程序代码大全.pdf
- 单片机数字时钟资料.doc
- 11项目管理前沿1.0.pptx
- 基于ssm的“魅力”繁峙宣传网站的设计与实现论文.doc
- 智慧交通综合解决方案.pptx
- 建筑防潮设计-PowerPointPresentati.pptx
- SPC统计过程控制程序.pptx
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈



评论5