多祖先荟萃分析转录组关联研究的最佳实践:全球生物库的图形摘要
PDF格式 | 1.31MB |
更新于2025-01-16
| 105 浏览量 | 举报

短文
多祖先、荟萃分析转录组关联研究的最佳实践:
来自全球生物库荟萃分析倡议的
图形摘要
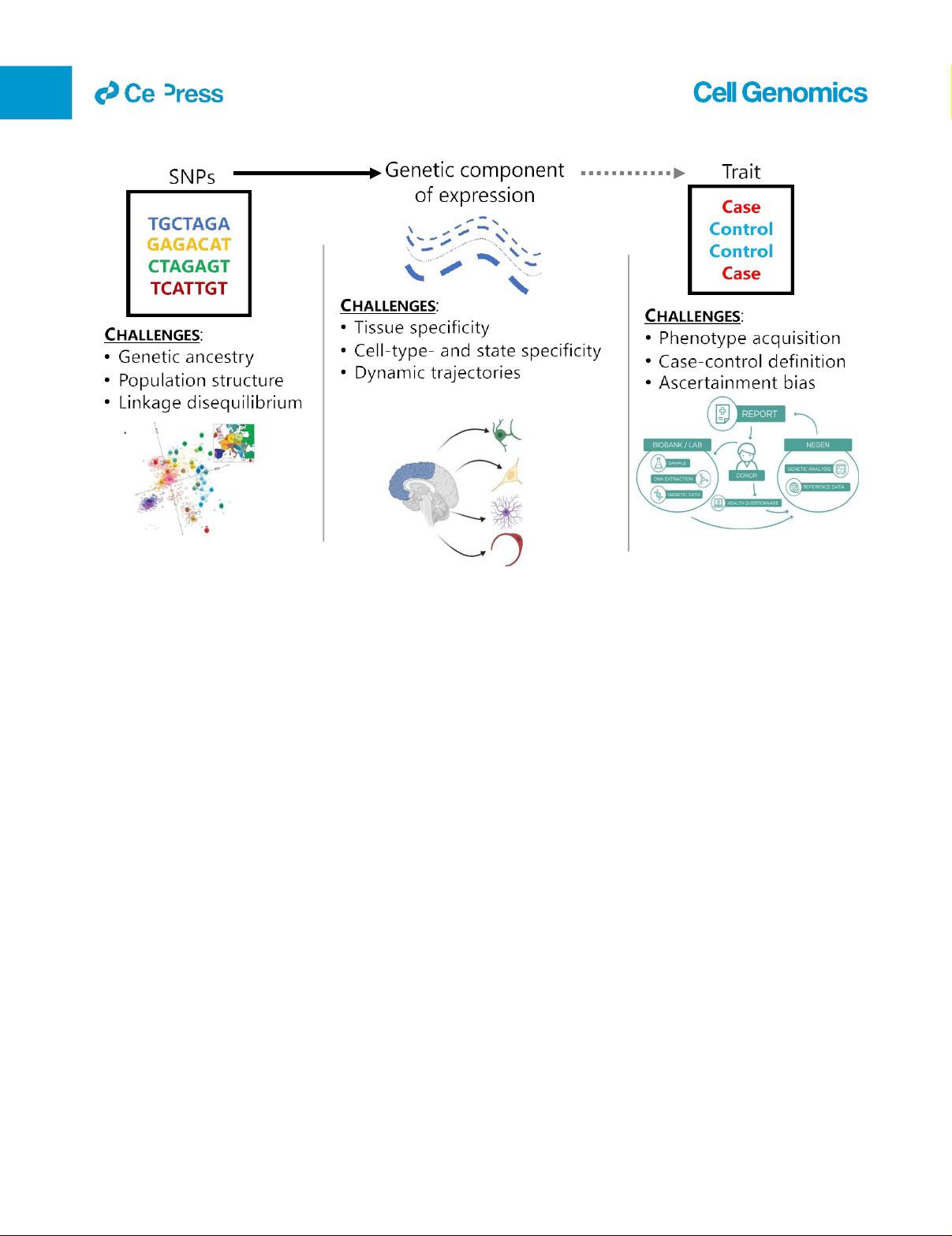
亮点
d
表达预测和基因
-
性状关联不跨祖先群体
d
建议对血统和研究进行荟萃分析,以减少检验统计量膨胀
d
详细的机械测试,以将
TWAS
的结果置于背景中
作者
Arjun Bhattacharya
,
Jibril B.
赫
伯,周丹,
...
,作者:
Eric R.
南
希
·
加马松
Cox
对应
abtbhatt@ucla.edu
简言之
Bhattacharya
和
Hirbo
等人描述了一个框
架,通过使用全转录组关联研究
(
TWAS
)对来自生物库的多祖先数据集
进行荟萃分析,确定基因水平与复杂性
状的遗传关联。他们展示了
TWAS
的陷
阱,并回顾了开放的方法学挑战,包括
缺乏表达预测的可移植性和结果的组织特
异性。
Bhattacharya等人,2022,细胞基因组学2,100180
2022
年
10
月
12
日
-
作者。
https://doi.org/10.1016/j.xgen.2022.100180
会


剩余15页未读,继续阅读
相关推荐




cpongm
- 粉丝: 6
 我的内容管理
展开
我的内容管理
展开







最新资源
- MATLAB实现ART与SART算法在医学CT重建中的应用
- S2SH整合版:快速搭建Struts2+Spring+Hibernate开发环境
- 托奇卡项目团队成员介绍
- 提升外链发布效率的SEO推广神器——搜易达网络推广大师v2.035
- C#打造简易记事本应用详细教程
- 探索虚拟现实地图VR的奥秘
- iOS模拟器屏幕截图新工具
- 深入解析JavaScript在生活应用开发中的运用
- STM32F10x函数库3.5中文版详解与应用
- 猎豹浏览器v6.0.114.13396 r1:安全防护与网购敢赔
- 掌握JS for循环输出的最简洁代码技巧
- Java入门教程:TranslationFileGenerator快速指南
- OpenDDS3.9源码解析及最新文档指南
- JavaScript提示框插件:鼠标滑过显示文章摘要
- MaskRCNN气球数据集:优质图像识别资源
- Laravel日志查看器:实现Apache多站点日志统一管理
