StringTie输出的转录本结果包括
时间: 2024-04-23 07:26:44 浏览: 17
StringTie输出的转录本结果包括以下信息:
1. 转录本ID:每个转录本都有一个唯一的ID,以“MSTRG”开头。
2. 基因ID:每个基因都有一个唯一的ID,以“MSTRG”开头。
3. 染色体:转录本所在的染色体。
4. 起始位置和终止位置:转录本的起始位置和终止位置。
5. 方向:转录本的方向,即正向还是反向。
6. 外显子:转录本包含的外显子的数量。
7. 转录本长度:转录本的长度。
8. 覆盖度:转录本的覆盖度,即转录本被RNA-Seq数据覆盖的程度。
9. FPKM:转录本的表达量,以FPKM为单位。
10. TPM:转录本的表达量,以TPM为单位。
11. 注释信息:转录本可能与已知的基因或转录本有重叠,因此可能包含注释信息。
注意:以上信息可能因不同的StringTie版本和参数设置而有所不同。
相关问题
stringtie怎么设置参数会输出转录本长度和外显子数量
在使用StringTie时,可以使用以下参数来输出转录本长度和外显子数量:
1. -G/--GTF:指定参考基因组文件,这个文件中包含了所有已知的转录本和基因的注释信息,以便StringTie能够比对和注释新的转录本。
2. -o/--output-dir:指定输出目录,这个目录中包含了所有的输出文件,包括注释文件和转录本表达量文件。
3. -e/--expr-counts:输出每个转录本的表达量和外显子数量,以及每个基因的表达量和外显子数量。
4. -B/--ballgown:输出Ballgown格式的转录本和基因表达量文件,这个文件中包含了每个样本的表达量和差异表达分析结果。
例如,使用以下命令行可以输出每个转录本的长度和外显子数量:
```
stringtie input.bam -G reference.gtf -e -o output.gtf
```
其中,input.bam是RNA-seq数据文件,reference.gtf是参考基因组文件,-e参数表示输出每个转录本的表达量和外显子数量,-o参数指定输出文件名为output.gtf。执行完毕后,output.gtf文件中将包含每个转录本的长度和外显子数量等信息。
tbtools提取最长转录本
tbtools是一个用于生物信息学分析的工具,可以用来提取基因组中的最长转录本。以下是使用tbtools提取最长转录本的步骤:
1. 打开tbtools软件。
2. 导入基因组数据。可以通过点击菜单栏中的"File",然后选择"Open"来导入基因组数据文件。
3. 在tbtools的菜单栏中选择"Gene",然后选择"Longest Transcript"。
4. 在弹出的窗口中,选择你想要提取最长转录本的基因组数据。
5. 点击"OK"按钮开始提取最长转录本。
6. 提取完成后,你可以在tbtools的结果窗口中查看提取的最长转录本。
请注意,以上步骤仅适用于使用tbtools软件提取最长转录本的情况。如果你使用其他工具或编程语言进行最长转录本的提取,步骤可能会有所不同。
相关推荐
最新推荐
无线语音遥控智能车.doc
无线语音遥控智能车
婚礼GO网站创业计划书.docx
"婚礼GO网站创业计划书"
在创建婚礼GO网站的创业计划书中,创业者首先阐述了企业的核心业务——GO婚礼设计,专注于提供计算机软件销售和技术开发、技术服务,以及与婚礼相关的各种服务,如APP制作、网页设计、弱电工程安装等。企业类型被定义为服务类,涵盖了一系列与信息技术和婚礼策划相关的业务。
创业者的个人经历显示了他对行业的理解和投入。他曾在北京某科技公司工作,积累了吃苦耐劳的精神和实践经验。此外,他在大学期间担任班长,锻炼了团队管理和领导能力。他还参加了SYB创业培训班,系统地学习了创业意识、计划制定等关键技能。
市场评估部分,目标顾客定位为本地的结婚人群,特别是中等和中上收入者。根据数据显示,广州市内有14家婚庆公司,该企业预计能占据7%的市场份额。广州每年约有1万对新人结婚,公司目标接待200对新人,显示出明确的市场切入点和增长潜力。
市场营销计划是创业成功的关键。尽管文档中没有详细列出具体的营销策略,但可以推断,企业可能通过线上线下结合的方式,利用社交媒体、网络广告和本地推广活动来吸引目标客户。此外,提供高质量的技术解决方案和服务,以区别于竞争对手,可能是其市场差异化策略的一部分。
在组织结构方面,未详细说明,但可以预期包括了技术开发团队、销售与市场部门、客户服务和支持团队,以及可能的行政和财务部门。
在财务规划上,文档提到了固定资产和折旧、流动资金需求、销售收入预测、销售和成本计划以及现金流量计划。这表明创业者已经考虑了启动和运营的初期成本,以及未来12个月的收入预测,旨在确保企业的现金流稳定,并有可能享受政府对大学生初创企业的税收优惠政策。
总结来说,婚礼GO网站的创业计划书详尽地涵盖了企业概述、创业者背景、市场分析、营销策略、组织结构和财务规划等方面,为初创企业的成功奠定了坚实的基础。这份计划书显示了创业者对市场的深刻理解,以及对技术和婚礼行业的专业认识,有望在竞争激烈的婚庆市场中找到一席之地。
管理建模和仿真的文件
管理Boualem Benatallah引用此版本:布阿利姆·贝纳塔拉。管理建模和仿真。约瑟夫-傅立叶大学-格勒诺布尔第一大学,1996年。法语。NNT:电话:00345357HAL ID:电话:00345357https://theses.hal.science/tel-003453572008年12月9日提交HAL是一个多学科的开放存取档案馆,用于存放和传播科学研究论文,无论它们是否被公开。论文可以来自法国或国外的教学和研究机构,也可以来自公共或私人研究中心。L’archive ouverte pluridisciplinaire
【基础】图像的几何变换:缩放、旋转与翻转

# 2.1 图像缩放的理论基础
图像缩放是一种几何变换,它可以改变图像的大小,使其适合特定的显示或处理需求。图像缩放可以通过以下变换矩阵来实现:
```
S = [[sx, 0, 0],
[0, sy, 0],
[0, 0, 1]]
```
其中:
* `sx` 和 `sy` 分别是水平和垂直缩放因子。
* `sx > 1` 和 `sy > 1` 表示图像放大。
* `sx < 1` 和
字节跳动面试题java
字节跳动作为一家知名的互联网公司,在面试Java开发者时可能会关注以下几个方面的问题:
1. **基础技能**:Java语言的核心语法、异常处理、内存管理、集合框架、IO操作等是否熟练掌握。
2. **面向对象编程**:多态、封装、继承的理解和应用,可能会涉及设计模式的提问。
3. **并发编程**:Java并发API(synchronized、volatile、Future、ExecutorService等)的使用,以及对并发模型(线程池、并发容器等)的理解。
4. **框架知识**:Spring Boot、MyBatis、Redis等常用框架的原理和使用经验。
5. **数据库相
微信行业发展现状及未来行业发展趋势分析.docx
微信行业发展现状及未来行业发展趋势分析
微信作为移动互联网的基础设施,已经成为流量枢纽,月活跃账户达到10.4亿,同增10.9%,是全国用户量最多的手机App。微信的活跃账户从2012年起步月活用户仅为5900万人左右,伴随中国移动互联网进程的不断推进,微信的活跃账户一直维持稳步增长,在2014-2017年年末分别达到5亿月活、6.97亿月活、8.89亿月活和9.89亿月活。
微信月活发展历程显示,微信的用户数量增长已经开始呈现乏力趋势。微信在2018年3月日活达到6.89亿人,同比增长5.5%,环比上个月增长1.7%。微信的日活同比增速下滑至20%以下,并在2017年年底下滑至7.7%左右。微信DAU/MAU的比例也一直较为稳定,从2016年以来一直维持75%-80%左右的比例,用户的粘性极强,继续提升的空间并不大。
微信作为流量枢纽,已经成为移动互联网的基础设施,月活跃账户达到10.4亿,同增10.9%,是全国用户量最多的手机App。微信的活跃账户从2012年起步月活用户仅为5900万人左右,伴随中国移动互联网进程的不断推进,微信的活跃账户一直维持稳步增长,在2014-2017年年末分别达到5亿月活、6.97亿月活、8.89亿月活和9.89亿月活。
微信的用户数量增长已经开始呈现乏力趋势,这是因为微信自身也在重新寻求新的增长点。微信日活发展历程显示,微信的用户数量增长已经开始呈现乏力趋势。微信在2018年3月日活达到6.89亿人,同比增长5.5%,环比上个月增长1.7%。微信的日活同比增速下滑至20%以下,并在2017年年底下滑至7.7%左右。
微信DAU/MAU的比例也一直较为稳定,从2016年以来一直维持75%-80%左右的比例,用户的粘性极强,继续提升的空间并不大。因此,在整体用户数量开始触达天花板的时候,微信自身也在重新寻求新的增长点。
中国的整体移动互联网人均单日使用时长已经较高水平。18Q1中国移动互联网的月度总时长达到了77千亿分钟,环比17Q4增长了14%,单人日均使用时长达到了273分钟,环比17Q4增长了15%。而根据抽样统计,社交始终占据用户时长的最大一部分。2018年3月份,社交软件占据移动互联网35%左右的时长,相比2015年减少了约10pct,但仍然是移动互联网当中最大的时长占据者。
争夺社交软件份额的主要系娱乐类App,目前占比达到约32%左右。移动端的流量时长分布远比PC端更加集中,通常认为“搜索下載”和“网站导航”为PC时代的流量枢纽,但根据统计,搜索的用户量约为4.5亿,为各类应用最高,但其时长占比约为5%左右,落后于网络视频的13%左右位于第二名。PC时代的网络社交时长占比约为4%-5%,基本与搜索相当,但其流量分发能力远弱于搜索。
微信作为移动互联网的基础设施,已经成为流量枢纽,月活跃账户达到10.4亿,同增10.9%,是全国用户量最多的手机App。微信的活跃账户从2012年起步月活用户仅为5900万人左右,伴随中国移动互联网进程的不断推进,微信的活跃账户一直维持稳步增长,在2014-2017年年末分别达到5亿月活、6.97亿月活、8.89亿月活和9.89亿月活。
微信的用户数量增长已经开始呈现乏力趋势,这是因为微信自身也在重新寻求新的增长点。微信日活发展历程显示,微信的用户数量增长已经开始呈现乏力趋势。微信在2018年3月日活达到6.89亿人,同比增长5.5%,环比上个月增长1.7%。微信的日活同比增速下滑至20%以下,并在2017年年底下滑至7.7%左右。
微信DAU/MAU的比例也一直较为稳定,从2016年以来一直维持75%-80%左右的比例,用户的粘性极强,继续提升的空间并不大。因此,在整体用户数量开始触达天花板的时候,微信自身也在重新寻求新的增长点。
微信作为移动互联网的基础设施,已经成为流量枢纽,月活跃账户达到10.4亿,同增10.9%,是全国用户量最多的手机App。微信的活跃账户从2012年起步月活用户仅为5900万人左右,伴随中国移动互联网进程的不断推进,微信的活跃账户一直维持稳步增长,在2014-2017年年末分别达到5亿月活、6.97亿月活、8.89亿月活和9.89亿月活。
微信的用户数量增长已经开始呈现乏力趋势,这是因为微信自身也在重新寻求新的增长点。微信日活发展历程显示,微信的用户数量增长已经开始呈现乏力趋势。微信在2018年3月日活达到6.89亿人,同比增长5.5%,环比上个月增长1.7%。微信的日活同比增速下滑至20%以下,并在2017年年底下滑至7.7%左右。
微信DAU/MAU的比例也一直较为稳定,从2016年以来一直维持75%-80%左右的比例,用户的粘性极强,继续提升的空间并不大。因此,在整体用户数量开始触达天花板的时候,微信自身也在重新寻求新的增长点。
微信作为移动互联网的基础设施,已经成为流量枢纽,月活跃账户达到10.4亿,同增10.9%,是全国用户量最多的手机App。微信的活跃账户从2012年起步月活用户仅为5900万人左右,伴随中国移动互联网进程的不断推进,微信的活跃账户一直维持稳步增长,在2014-2017年年末分别达到5亿月活、6.97亿月活、8.89亿月活和9.89亿月活。
微信的用户数量增长已经开始呈现乏力趋势,这是因为微信自身也在重新寻求新的增长点。微信日活发展历程显示,微信的用户数量增长已经开始呈现乏力趋势。微信在2018年3月日活达到6.89亿人,同比增长5.5%,环比上个月增长1.7%。微信的日活同比增速下滑至20%以下,并在2017年年底下滑至7.7%左右。
微信DAU/MAU的比例也一直较为稳定,从2016年以来一直维持75%-80%左右的比例,用户的粘性极强,继续提升的空间并不大。因此,在整体用户数量开始触达天花板的时候,微信自身也在重新寻求新的增长点。
"互动学习:行动中的多样性与论文攻读经历"
多样性她- 事实上SCI NCES你的时间表ECOLEDO C Tora SC和NCESPOUR l’Ingén学习互动,互动学习以行动为中心的强化学习学会互动,互动学习,以行动为中心的强化学习计算机科学博士论文于2021年9月28日在Villeneuve d'Asq公开支持马修·瑟林评审团主席法布里斯·勒菲弗尔阿维尼翁大学教授论文指导奥利维尔·皮耶昆谷歌研究教授:智囊团论文联合主任菲利普·普雷教授,大学。里尔/CRISTAL/因里亚报告员奥利维耶·西格德索邦大学报告员卢多维奇·德诺耶教授,Facebook /索邦大学审查员越南圣迈IMT Atlantic高级讲师邀请弗洛里安·斯特鲁布博士,Deepmind对于那些及时看到自己错误的人...3谢谢你首先,我要感谢我的两位博士生导师Olivier和Philippe。奥利维尔,"站在巨人的肩膀上"这句话对你来说完全有意义了。从科学上讲,你知道在这篇论文的(许多)错误中,你是我可以依
【基础】OpenCV中的基本图像操作
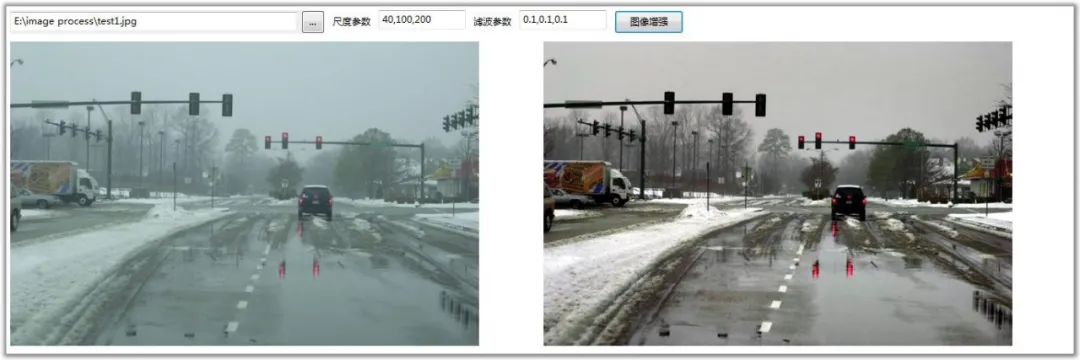
# 2.1 图像的基本概念和表示
### 2.1.1 图像的像素和颜色空间
图像由像素组成,每个像素表示图像中一个点的颜色和亮度信息。像素的排列方式决定了图像的形状和大小。
颜色空间定义了表示图像中颜色的方式。常用的颜色空间包括 RGB(红色、绿色、蓝色)、HSV(色调、饱和度、明度)和 YUV(亮度、色度)。不同的颜色空间适用于不同的图像处理任务。
### 2.1.2 图像的存储和加载
图像可以
# 请根据注释在下面补充你的代码实现knn算法的过程 # ********** Begin ********** # # 对ndarray数组进行遍历
K-Nearest Neighbors (KNN) 算法是一种基于实例的学习方法,用于分类和回归分析。在代码中,实现KNN的基本步骤如下:
```python
# 导入必要的库
import numpy as np
from collections import Counter
# 假设我们有一个训练数据集 X_train 和对应的标签 y_train
X_train = ... # (n_samples, n_features)
y_train = ... # (n_samples)
# KNN函数实现
def knn_k(X_test, k, X_train, y_train):
信息技术与教育.docx
信息技术与教育是一个关键领域,它探讨了如何有效地将计算机科学(CS)技术融入教育体系,提升教学质量和学习体验。以下是关于该主题的一些重要知识点:
1. **逻辑“与”检索**:在信息检索中,逻辑“与”操作用于同时满足多个条件的查询,确保结果包含所有指定的关键词,提高搜索的精确度。
2. **通配符“*”的应用**:通配符“*”(星号)在搜索中代表任意字符序列,帮助用户查找类似或部分匹配的关键词,扩大搜索范围。
3. **进阶搜索引擎检索技巧**:理解并运用高级搜索选项,如布尔运算、过滤器和自定义排序,能够更高效地筛选和分析搜索结果。
4. **教育目标与编写方法**:B选项对应的学习目标可能是具体的教学策略或技能,可能是指将信息技术融入课程设计中的具体步骤。
5. **课程整合与变革**:将信息技术融入课程整体,涉及课程内容和结构的创新,这是支持教育变革的一种观点。
6. **经验之塔理论**:该理论区分了从实践操作到抽象概念的认知层次,电影与电视在经验之塔中处于较为具体的底层经验。
7. **信息素养的侧重点**:信息能力被认为是信息素养的重点与核心,强调个体获取、评估、管理和创造信息的能力。
8. **教学评价类型**:学习过程中可以进行过程性评价和总结性评价,前者关注学习过程,后者评估最终成果。
9. **网络课程的支撑**:网络及通讯技术为网络课程提供了基础设施和环境支持,确保在线学习的顺利进行。
10. **PowerPoint演示模式**:演讲者模式允许演讲者在幻灯片展示的同时查看备注,增强讲解的灵活性。
11. **“经验之塔”层级**:电影与电视作为视听媒体,对应的是相对具体的实践经验,位于经验之塔的较低层。
12. **教育信息化的兴起**:20世纪90年代,伴随“全国学习网”等项目的建设,教育信息化的概念逐渐被提出。
13. **信息技术与课程整合误区**:错误的做法包括认为存在固定模式,以及忽视信息技术作为学生主动学习工具的角色。
14. **先行组织者教学策略**:由美国心理学家George A. Bormann提出的教学策略,用于引导学生理解和准备新知识。
15. **校本教研方式**:D选项可能是非主要的校本教研方式,通常包括同伴互助、专业发展研讨会等形式。
16. **信息化教育的核心**:信息化教育的核心是教育信息资源的利用和整合,促进教育质量的提升。
17. **信息技术与科研任务整合模式**:学生通过信息技术完成科研任务,体现的是信息技术作为学习工具和科研支持的作用。
18. **中国知网资源下载**:中国知网除了CAJ格式,还提供PDF或其他格式的资源下载。
19. **多媒体课件编辑软件**:Authorware是一种常用的多媒体课件制作工具,Windows不是编辑软件。
20. **问题设计原则**:问题设计需要具有一定的复杂性和歧义性,以激发思考和批判性思维。
21. **学习理论基础**:学习被解释为刺激与反应之间的联结,但这里的“及时强化”可能指的是行为主义学习理论中的强化机制。
以上是关于信息技术与教育的一些关键知识点,涵盖了课程设计、信息检索、教育理论与实践等方面。