【NAMD模拟前处理艺术】:系统构建、能量最小化与热平衡的实战技巧
发布时间: 2024-12-16 16:08:46 阅读量: 2 订阅数: 5 

nix-container-namd2.14-cuda10.2:使用Nix的CUDA 10.2支持的NAMD 2.14容器

参考资源链接:[NAMD分子动力学模拟教程:从入门到进阶分析](https://wenku.csdn.net/doc/845t0u7fv4?spm=1055.2635.3001.10343)
# 1. NAMD模拟前处理艺术概览
在生物分子模拟的领域中,NAMD(NAnoscale Molecular Dynamics)作为一种先进的模拟软件,被广泛应用于生命科学和材料科学的研究。本章将概述NAMD模拟前处理的基本步骤和要点,为读者提供一个清晰的路径,以准备和优化分子动力学模拟的参数。
## 1.1 NAMD模拟前处理的重要性
分子动力学模拟能够帮助我们理解分子在时间尺度上的动态行为。在模拟开始之前,进行周密的前处理工作,是确保模拟结果准确性和有效性的关键。前处理涉及到创建初始结构、定义力场参数、设定合理的模拟盒子、溶剂化模型和离子化条件等。
## 1.2 前处理的艺术:关键步骤
前处理的艺术包括如下关键步骤:
- **选择合适的力场**:力场是模拟中描述原子间相互作用的数学模型,选择一个合适的力场对于获得准确的模拟结果至关重要。
- **初始化模型设置**:包括定义模拟盒子的大小、形状,以及构建适当的溶剂化环境。
- **优化系统结构**:通过一系列预模拟步骤,如能量最小化和热平衡,来达到一个稳定的状态,为后续的生产模拟做准备。
通过以上内容,我们可以看到,NAMD模拟前处理的每一步都涉及到复杂的决策和细致的操作,本章的后续内容将逐一深入探讨这些步骤,为读者提供在NAMD模拟前处理中的实际指导。
# 2. 系统构建的策略与实践
构建一个精确的系统模型是分子动力学(MD)模拟成功的关键。这一过程涉及从选择合适的建模工具开始,通过组装、优化,直至验证最终的系统模型。系统构建不仅要求我们对分子模型有深入的理解,同时也要求我们具备相关软件工具的熟练操作能力。
## 2.1 分子建模基础
分子建模是分子动力学模拟中的第一步,也是至关重要的一步。建模的质量直接影响到模拟结果的准确性和可靠性。
### 2.1.1 原子和分子的表示方法
在分子建模中,原子和分子通常通过坐标和键类型来表示。原子的位置由三个空间坐标(x, y, z)表示,而分子间作用力则由键、键角和二面角来定义。其中,键类型包括共价键、范德华力、氢键等。
### 2.1.2 建模工具的选择与使用
选择合适的建模工具对于构建高质量的分子系统至关重要。常用的建模工具有GROMACS、CHARMM、NAMD等。以NAMD为例,它通过其自身的配置文件`.pdb`和`.psf`文件来分别定义原子的位置和拓扑结构。以下是使用NAMD建立模型的基本步骤:
1. 准备蛋白质结构文件(PDB格式)。
2. 生成拓扑文件(PSF格式),这通常需要使用VMD或CHARMM工具。
3. 进行能量最小化处理,以修正模型的构象。
4. 准备模拟盒子并添加溶剂模型(如TIP3P、SPC/E等)。
5. 定义周期性边界条件和离子化环境。
```bash
# 示例:生成拓扑文件的命令
vmd -dispdev text -e psfgen.tcl
```
在这个例子中,`psfgen.tcl`是一个包含了生成拓扑文件步骤的Tcl脚本。
## 2.2 系统组装技巧
组装系统包括晶胞构建、扩展、溶剂添加和离子化处理,这是确保模拟盒子正确和有效的重要步骤。
### 2.2.1 晶胞构建与扩展
晶胞是模拟系统的基础结构,它可以是简单立方体、面心立方体等。构建和扩展晶胞是为了容纳蛋白质、溶剂和离子,以便模拟自然环境中的相互作用。操作过程中需要考虑蛋白质与晶胞边界间的距离,通常至少为1nm,以避免边界效应。
### 2.2.2 溶剂添加与离子化处理
溶剂是模拟生物分子生存环境的必要组成部分,常用的溶剂模型包括TIP3P、SPC/E等。添加溶剂后,为了保持系统的电中性,需要添加相应的反离子。这一步骤通常涉及到NAMD的离子化插件或VMD脚本。
## 2.3 模型验证与优化
构建的模型需要经过严格验证和优化,以确保其生物物理学的真实性。模型验证主要关注原子的类型、数量以及拓扑结构的准确性。
### 2.3.1 模型质量检查标准
检查模型的质量通常涉及以下几个方面:
- **完整性检查**:确保所有的原子和残基都被正确地包含在拓扑文件中。
- **几何验证**:通过检查键长、键角、二面角来评估模型的几何结构。
- **能量检查**:评估系统中的非键相互作用,如范德华力和库仑力,以确认潜在的异常。
```python
import MDAnalysis as mda
# 示例:使用MDAnalysis检查蛋白质结构的质量
u = mda.Universe('protein.pdb', 'topology.psf')
print(u.select_atoms('protein').n_residues)
```
在此示例中,`MDAnalysis`库用于检查蛋白质中残基的数量,帮助评估拓扑文件的完整性。
### 2.3.2 缺陷修正与能量优化
即使经过了仔细的检查,模型中仍然可能存在一些缺陷,如不当的原子类型或不合适的键定义等。修正这些缺陷后,还需要对整个系统进行能量最小化处理,以去除过高的能量和避免潜在的结构冲突。
```bash
# 示例:NAMD
```

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏目录
最低0.47元/天 解锁专栏
买1年送1年
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
故障无处遁形:PJLink 1.04 Class1故障排除与性能提升秘籍

参考资源链接:[PJLink 1.04协议:简化多设备网络投影机控制](https://wenku.csdn.net/doc/6412b761be7fbd1778d4a186?spm=1055.2635.3001.10343)
# 1. PJLink 1.04 Class1简介与故障
【故障排除实战指南】:iFIX连接SQL数据库的常见问题与解决捷径

参考资源链接:[IFIX与SQL数据库连接及IFIX2DB工具使用教程](https://wenku.csdn.net/doc/6412b7
【PADS Router元件库管理全攻略】:维护和优化,提升设计灵活性

参考资源链接:[PADS Router全方位教程:从布局到高速布线](https://wenku.csdn.net/doc/1w7vayrbdc?spm=1055.2635.3001.10343)
# 1. PADS Router元件库基础与重要性
在现代电路板设计中,元件库是支撑设计高效运行的
【破解DAHUA HTTP API】:5个必知的请求与响应技巧
参考资源链接:[大华官方2018-11版HTTP接口协议CGI规范与安全建议](https://wenku.csdn.net/doc/6412b6dcbe7fbd1778d483d5?spm=1055.2635.3001.10343)
# 1. DAHUA HTTP API概述
在现代的信息技术生态中,API(应用程序编程接口)已经成为应用间沟通的桥梁。DAHUA,作为一家知名
微信小程序触摸反馈的用户交互指南:如何设计更吸引人的反馈
参考资源链接:[微信小程序滑动翻页效果实现教程](https://wenku.csdn.net/doc/6459ff3bfcc5391368262691?spm=1055.2635.3001.10343)
# 1. 微信小程序触摸反馈的重要性
在当今的移动互联网时代,微信小程序已经成为企业和开发者进入市场的重要途径。用户与小程序的交互过程中,触摸反馈发挥着至关重
数字信号处理快速学习法:PPT课件带你速成基础知识
参考资源链接:[数字信号处理(第三版)PPT课件](https://wenku.csdn.net/doc/645f4789543f8444888b11a3?spm=1055.2635.3001.10343)
# 1. 数字信号处理概述
数字信号处理(Digital Signal Processing, DSP)是电子工程领域中一个非常重要的分支,它主要研究如何通过数字计算手段对信号进行分析和处理
【机械设计中的花键力量】:DIN 5480标准与现代机械的融合

参考资源链接:[DIN 5480: 渐开线花键技术规范详解](https://wenku.csdn.net/doc/6k18cpv1qq?spm=1055.2635.3001.10343)
# 1. 花键及其在机械设计中的重要性
## 1.1 花键的定义与特点
花键是一种机械传动元件,通常用于连接轴与齿轮、滑轮等旋转部件。与键槽和紧定螺钉相比,花键具有更
三菱PLC与台达VFD-L通讯监控:构建可视化管理界面的秘诀
参考资源链接:[三菱PLC与台达VFD-L变频器RS485通讯详解及设置](https://wenku.csdn.net/doc/6451ca45ea0840391e7382a7?spm=1055.2635.3001.10343)
# 1. 三菱PLC与台达VFD-L通讯概述
在现代工业自动化系统中,三菱PLC(可编程逻辑控制器)与台
【SIMCA 14生物统计学应用】:生命科学数据分析,信手拈来
参考资源链接:[SIMCA 14 用户手册:全方位数据分析指南](https://wenku.csdn.net/doc/3f5cnjutvk?spm=1055.2635.3001.10343)
# 1. SIMCA 14生物统计学应用概览
在本章中,我们将简要介绍SIMCA 14软件在生物统计学应用中的重要性与作用。SIMCA(Soft Independent Modeling of Class Anal
CMOS版图设计抗噪声艺术:非门与或门的稳健设计方法
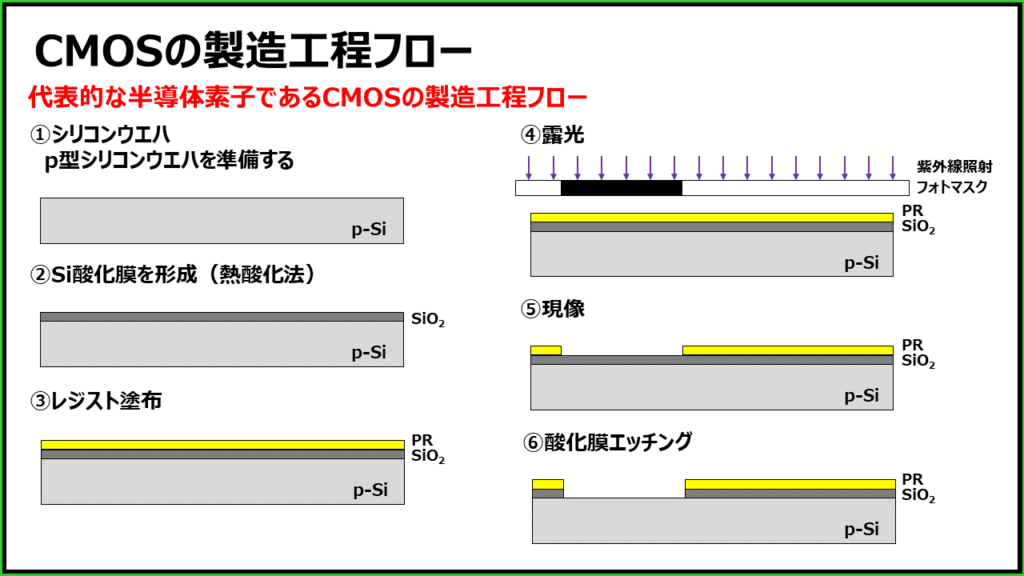
参考资源链接:[掌握CMOS与非/或非门版图设计:原理图与仿真实战](https://wenku.csdn.net/doc/4f6w6qtz7b?spm=1055.2635.3001.10343)
# 1. CMOS版图设计概述与噪声问题
## 1.1 版图设计基础
CMOS(互补金属氧化物半导体)技术是现代集成电路设计的基石,其中版图设计是实现芯片功
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送1年
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )