【蛋白质折叠动力学分析】:NAMD案例研究详解
发布时间: 2024-12-16 15:54:14 阅读量: 4 订阅数: 5 

分子动力学分析软件namd 2.7
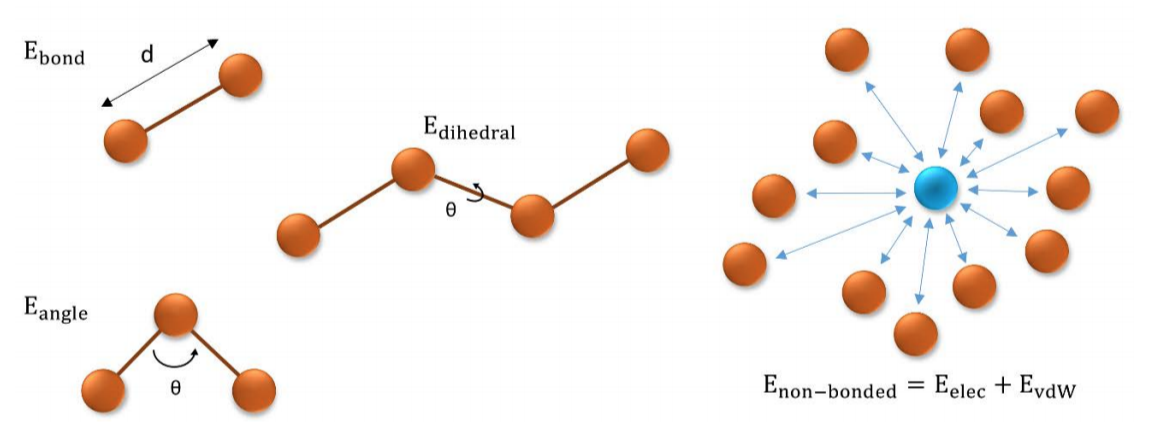
参考资源链接:[NAMD分子动力学模拟教程:从入门到进阶分析](https://wenku.csdn.net/doc/845t0u7fv4?spm=1055.2635.3001.10343)
# 1. 蛋白质折叠动力学研究概述
## 1.1 蛋白质折叠的重要性
蛋白质折叠是生物学研究的核心问题之一,其过程和机制关系到生物体内许多生化活动。正确折叠的蛋白质是生命活动不可或缺的基础,而错误的折叠则可能导致多种疾病。科学家们通过研究蛋白质折叠的动力学,不仅可以揭示蛋白质如何在极短的时间内完成折叠,还能在理论上预测其三维结构和功能,为治疗相关疾病提供科学依据。
## 1.2 动力学研究的现代技术
随着计算生物学的发展,分子动力学模拟已经成为研究蛋白质折叠动力学的重要手段。通过模拟蛋白质在分子层面的行为,研究人员能够在原子水平上观察蛋白质的折叠过程。NAMD模拟工具作为其中的佼佼者,因其处理能力强大、应用灵活广泛而受到广泛关注。本系列文章将从NAMD的安装、应用到蛋白质折叠动力学的深入理解,全面介绍如何利用NAMD进行蛋白质研究。
# 2. NAMD模拟工具的基础应用
### 2.1 NAMD简介和安装配置
#### 2.1.1 NAMD的发展历程和功能特点
NAMD,全称是NAnoscale Molecular Dynamics,是一个专门为大规模分子动力学模拟设计的程序。由Theoretical and Computational Biophysics Group在伊利诺伊大学厄巴纳-香槟分校开发。NAMD的第一个版本发布于1995年,经过几十年的发展和优化,现在已经是分子生物学和生物物理学领域应用最广泛的模拟工具之一。
NAMD的一个显著特点是它采用了并行计算的架构,可以在多核处理器以及分布式内存计算集群上实现高效运算。它支持多种复杂的生物分子系统,包括蛋白质、核酸、糖类和脂类,并能模拟它们在各种环境条件下的行为。NAMD通过 CHARMM(Chemistry at HARvard Molecular Mechanics)力场等参数,实现了对生物分子的精确描述。
#### 2.1.2 安装NAMD所需的系统环境和依赖
为了使用NAMD进行分子动力学模拟,用户需要准备具备特定配置的操作系统环境。以下是NAMD安装前必须满足的基本要求:
- 操作系统:支持Linux和Mac OS X,部分版本支持Windows(通过Cygwin或者虚拟机)。
- 编译器:需要GCC或者Intel编译器。
- 并行环境:MPICH或OpenMPI。
- 依赖库:zlib, libjpeg(用于可视化模块)。
安装步骤通常包括下载预编译的二进制文件或从源代码编译。在Linux系统中,推荐使用包管理器安装依赖,例如在基于Debian的系统中可以使用以下命令:
```bash
sudo apt-get install build-essential openmpi-bin libopenmpi-dev libfftw3-dev zlib1g-dev libjpeg-dev
```
在安装了必要的依赖后,用户可以从NAMD官方网站下载相应的二进制文件包或者源代码进行安装。NAMD社区还提供了详细的安装指南和脚本,可以帮助用户顺利完成安装。
### 2.2 NAMD的基本命令和操作
#### 2.2.1 NAMD输入文件的构成和编写
NAMD模拟的执行依赖于输入文件的编写,输入文件通常包含模拟的参数设置,结构文件的路径,以及其他相关的模拟指令。一个基础的NAMD输入文件包含了以下部分:
- **Structure:** 指定蛋白质结构文件。
- **Coordinates:** 指定初始坐标。
- **Temperature:** 设置系统的初始温度。
- **Pressure:** 设置系统的压力。
- **Periodic Boundary Conditions (PBC):** 设置周期边界条件。
- **Minimization:** 最小化能量。
- **Equilibration:** 系统平衡。
- **Production:** 生产性模拟。
- **Output:** 设置输出频率和输出参数。
这里是一个简单的NAMD输入文件示例:
```tcl
# NAMD configuration file for a simple protein simulation
structure protein.psf
coordinates protein.pdb
temperature 300
seed 12345
ensemble NVT
timestep 1
exclude scaled1-4
1-4scaling 0.833333
cutoff 12
switching on
switchdist 10
pairlistdist 13.5
margin 2
langevin on
langevinTemp 300
langevinHydrogen no
numsteps 1000
outputName output
```
#### 2.2.2 运行NAMD模拟的基本流程
在有了正确的输入文件之后,可以使用NAMD命令行工具来启动模拟。一个简单的NAMD运行命令如下:
```bash
namd2 config.namd
```
模拟运行时,NAMD会在当前目录下生成一系列输出文件,包括日志文件、能量文件、坐标文件等。模拟完成后,这些文件可用于分析模拟数据。
#### 2.2.3 常用的NAMD模拟参数设置
在NAMD输入文件中,用户可以根据需要设置多种模拟参数以适应特定的研究目标。以下是一些常用的模拟参数设置:
- **结构文件(structure):** 指定蛋白质的拓扑文件(PSF格式)。
- **坐标文件(coordinates):** 提供蛋白质的原子坐标(PDB格式)。
- **温度(temperature):** 指定模拟的温度值。
- **压力(pressure):** 指定模拟的压力值,通常用作NPT系综。
- **ensemble:** 指定模拟的系综类型,如NVT或NPT。
- **timestep:** 设置模拟的时间步长,影响模拟速度和准确性。
- **exclude:** 设置用于计算非键相互作用的排除区域。
- **cutoff:** 设置非键相互作用的截断距离。
- **langevin:** 启用Langevin动力学,用于温度控制。
- **numsteps:** 指定模拟总步数。
这些参数设置对模拟结果有显著影响,因此在模拟开始前,需要根据具体研究目标仔细选择。
### 2.3 NAMD与生物信息学数据的整合
#### 2.3.1 蛋白质结构文件的处理和转换
在NAMD模拟之前,通常需要将蛋白质的结构文件从一种格式转换为适用于NAMD的PSF格式。常用的蛋白质结构文件格式有PDB(蛋白质数据银行格式),这个格式广泛用于存储生物大分子的三维结构信息。
PDB文件通过文本格式保存了原子坐标、残基类型和键连接信息。为了将PDB文件转换为PSF文件,可以使用VMD(Visual Molecular Dynamics)软件。VMD不仅能进行分子模型的可视化,还能进行分子结构的编辑和转换。以下是使用VMD进行文件转换的基本步骤:
1. 打开VMD并导入PDB文件。
2. 在VMD中使用`mol`命令创建分子对象。
3. 使用`psfgen`插件生成PSF文件。
#### 2.3.2 将实验数据融入NAMD模拟的方法
将实验数据融入NAMD模拟,可以提高模拟结果的准确性和可靠性。常见的实验数据类型包括X射线晶体学数据、NMR数据以及冷冻电镜数据等。以下是几种将实验数据融入NAMD模拟的方法:
- **距离限制(Distance Restraints):** 通过对某些原子对之间的距离施加限制,模拟实验数据中观察到的距离。
- **角度限制(Angle Restraints):** 类似地,限制原子间的角度,以符合实验数据。
- **位置限制(Position Restraints):** 限制某些原子在空间中的位置,使模拟系统与实验结构保持一致。
整合这些实验数据的方法通常涉及到在NAMD输入文件中设置适当的参数。下面是一个如何在NAMD输入文件中设置距离限制的示例:
```tcl
# NAMD input file with distance restraints
fixedAtoms on
fixedAtomsFile fixedatoms.pdb
fixedAtomsCol B
set all [atomselect top all]
foreach atom [$all get index] {
if {$all($atom,restname) == "CA"} {
print "Setting distance restraint for residue $all($atom,residue)"
addgroup {1 2} ;#
```

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏目录
最低0.47元/天 解锁专栏
买1年送1年
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
数字信号处理系统设计指南:工程实现的PPT课件要点

参考资源链接:[数字信号处理(第三版)PPT课件](https://wenku.csdn.net/doc/645f4789543f8444888b11a3?spm=1055.2635.3001.10343)
# 1. 数字信号处理基础概念
## 1.1 什么是数字信号处理?
数字信号处理(Digital Signal Processing,简称DSP)是利用数字计算机
【SIMCA 14脚本编写基础】:自动化分析流程,省时省力又省心

参考资源链接:[SIMCA 14 用户手册:全方位数据分析指南](https://wenku.csdn.net/doc/3f5cnjutvk?spm=1055.2635.3001.1034
【CMOS版图设计最佳实践】:非门与或门布局走线技巧全攻略
参考资源链接:[掌握CMOS与非/或非门版图设计:原理图与仿真实战](https://wenku.csdn.net/doc/4f6w6qtz7b?spm=1055.2635.3001.10343)
# 1. CMOS版图设计基础
## 1.1 CMOS技术简介
CMOS(互补金属氧化物半导体)技术是现代集成电路设计中最重要的技术之一。它使用N型和P型两种类型的 MOSFET(金属氧化物半导体场效应晶体管)以互补的方式工作,从而实现低功耗和高效性能的集成电路。CMOS技术的这一特性,使其在制造成本、功耗、速度等方面具有明显优势,被广泛应用于各种数字电路、微处理器、存储器和模拟电路的设计中。
【精确检测与花键制造】:DIN 5480标准下的测量技术与方法
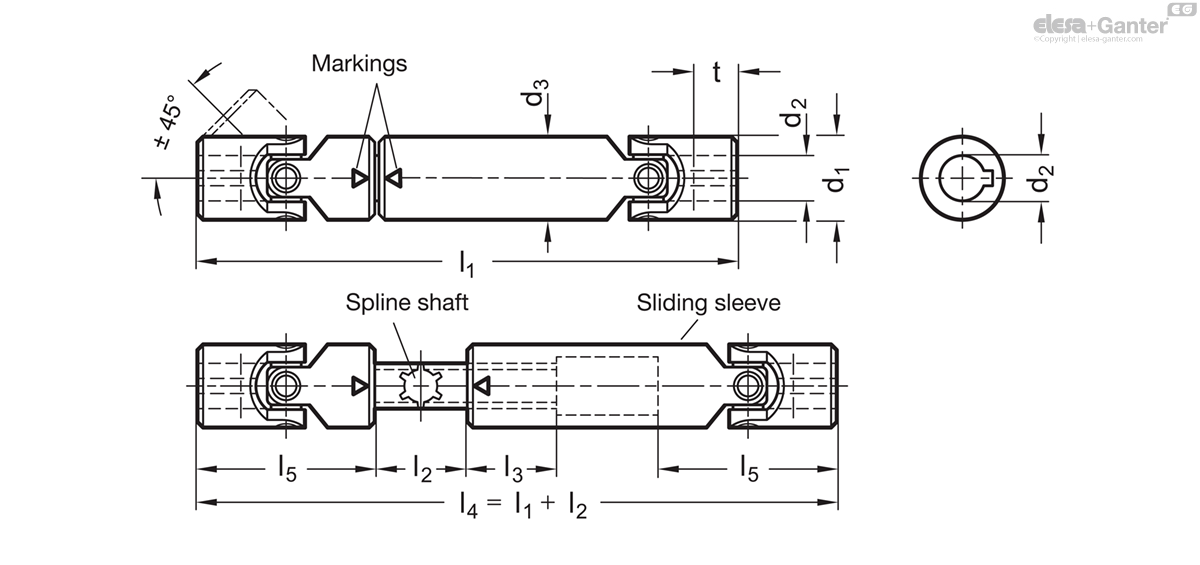
参考资源链接:[DIN 5480: 渐开线花键技术规范详解](https://wenku.csdn.net/doc/6k18cpv1qq?spm=1055.2635.3001.10343)
#
【PADS Router多层板设计攻略】:破解复杂电路板设计的秘诀

参考资源链接:[PADS Router全方位教程:从布局到高速布线](https://wenku.csdn.net/doc/1w7vayrbdc?spm=1055.2635.3001.10343)
# 1. PADS Router多层板设计概述
在电子工业迅猛发展的今天,多层板设计已成为现代
测试与调试的艺术:全数字锁相环中的Bang-Bang鉴相器精确控制(调试技巧大公开)

参考资源链接:[全数字锁相环设计:Bang-Bang鉴相器方法](https://wenku.csdn.net/d
微信小程序复杂界面流畅滑动解决方案:提升性能与体验

参考资源链接:[微信小程序滑动翻页效果实现教程](https://wenku.csdn.net/doc/6459ff3bfcc5391368262691?spm=1055.2635.3001.10343)
# 1. 微信小程序界面滑动性能的重要性
在当今的数字时代,用户体验已经成为衡量一款应用成功与否的重要指标之一。对于微信小程序而言,界面的滑动性能更是直
【数据同步深度解析】:iFIX与SQL数据库同步机制的全面揭秘
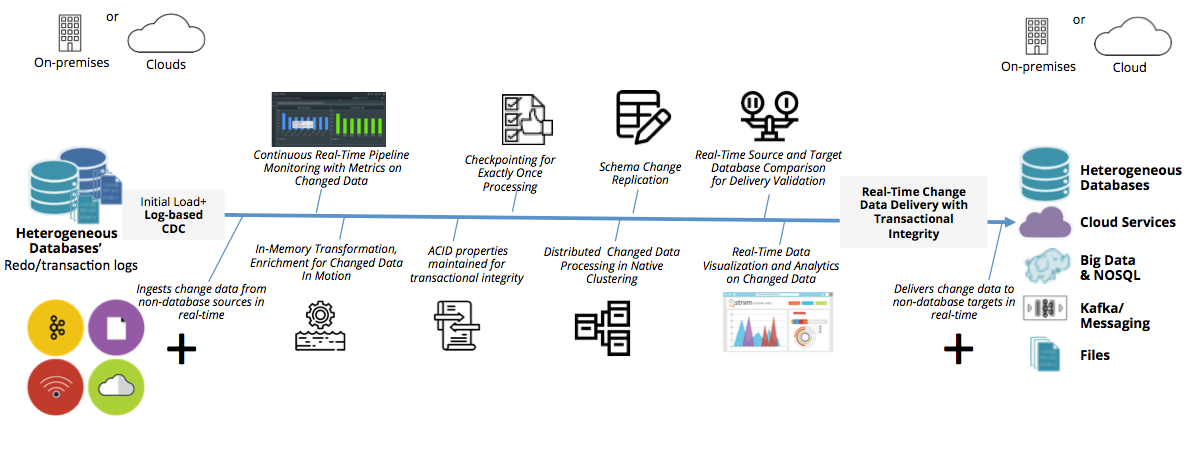
参考资源链接:[IFIX与SQL数据库连接及IFIX2DB工具使用教程](https://wenku.csdn.net/doc/6412b77cbe7fbd1778d4a76f?spm=1055.2635.3001.10343)
# 1. iFIX与SQL数据库同步概述
在信息技术的浪潮中,工业自动化领域的数据管理正在向更加动态和智能的方向发展。iF
三菱PLC与台达VFD-L通讯优化实战:稳定数据传输的5大策略

参考资源链接:[三菱PLC与台达VFD-L变频器RS485通讯详解及设置](https://wenku.csdn.net/doc/6451ca45ea0840391e7382a7?spm=1055.2635.3001.10343)
# 1. 三菱PLC与台达VFD-L通讯概述
在工业自动化领域,实现设备之间的有效通信是至关重要的。三菱PLC(可编程逻辑控制器)与台达VFD-L(
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送1年
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )