【Star CCM+化学反应模拟】:模拟化学过程,解锁现实世界的秘密
发布时间: 2024-12-02 22:52:12 阅读量: 6 订阅数: 13 

参考资源链接:[STAR-CCM+用户指南:版本13.02官方文档](https://wenku.csdn.net/doc/2x631xmp84?spm=1055.2635.3001.10343)
# 1. Star CCM+化学反应模拟概述
在现代化学工程与材料科学中,模拟工具已成为不可或缺的辅助手段,而Star CCM+作为一个功能强大的计算流体动力学(CFD)模拟软件,在化学反应模拟领域展现了巨大的潜力。通过模拟,我们可以预知反应条件变化对化学反应过程的影响,优化反应器设计,提高产物的纯度和产率。本章将对Star CCM+在化学反应模拟中的应用进行概述,并解释其在进行复杂化学反应模拟时的重要性。
在这一章中,我们将了解到Star CCM+如何帮助化学工程师理解和控制化学反应的过程,包括它在模拟流体流动、热量传递和质量传递时如何与化学反应动力学相结合。这些模拟不仅在实验室规模上进行验证,还能在工业生产规模上进行优化,显著降低研发成本和时间。对于初次接触Star CCM+的用户来说,本章提供了一个快速入门的平台,为进一步深入学习和应用打下基础。
# 2. 化学反应模拟的理论基础
## 2.1 化学动力学与反应速率
### 2.1.1 动力学方程的推导
化学动力学是研究化学反应速率以及反应过程的学科。在化学反应模拟中,动力学方程通常用来预测反应物质随时间的变化。首先,我们以一个简单的单分子反应为例,探讨其基本的动力学方程。
假设我们有一级反应 A → 产物,其动力学方程可以表示为:
\[ -\frac{d[A]}{dt} = k[A] \]
这里 \([A]\) 是反应物A的浓度,\(k\) 是反应速率常数,而 \(-\frac{d[A]}{dt}\) 表示随时间减少的A的速率。积分这个方程,我们可以得到:
\[ \ln \frac{[A]_0}{[A]} = kt \]
其中 \([A]_0\) 是初始浓度。通过这个方程,我们可以在给定时间 \(t\) 时计算出反应物A的剩余浓度。
### 2.1.2 反应速率的影响因素
反应速率受到多种因素的影响,如温度、浓度、压力、催化剂的存在以及反应物的表面积等。例如,根据阿伦尼乌斯方程:
\[ k = Ae^{-\frac{E_a}{RT}} \]
这里 \(A\) 是频率因子,\(E_a\) 是活化能,\(R\) 是气体常数,而 \(T\) 是绝对温度。从这个关系中,我们可以看出温度对反应速率的影响非常显著,通常温度的升高会显著增加反应速率。
## 2.2 化学平衡理论
### 2.2.1 平衡常数的理解和计算
化学平衡是指在一定条件下,正反两个方向的化学反应速率相等,反应系统中的物质组成不再发生变化的状态。对于一个可逆反应:
\[ aA + bB \rightleftharpoons cC + dD \]
化学平衡常数 \(K\) 的定义是:
\[ K = \frac{[C]^c[D]^d}{[A]^a[B]^b} \]
平衡常数 \(K\) 的值只取决于温度,与反应物和产物的初始浓度无关。通过测量平衡时各组分的浓度,我们可以计算出平衡常数。
### 2.2.2 影响化学平衡的因素
根据勒沙特列原理,如果改变平衡系统的条件之一(如温度、压力、浓度),平衡将向减弱这种改变的方向移动。例如,对于气体参与的反应,增加压力会导致生成物的摩尔数减少的方向移动,增加温度则会向吸热方向移动。
## 2.3 热力学在化学反应模拟中的应用
### 2.3.1 熵、焓与自由能的概念
热力学在化学反应模拟中起着至关重要的作用。熵 (\(S\)) 表示系统无序程度,焓 (\(H\)) 表示系统热含量,而自由能 (\(G\)) 是系统能量变化的热力学潜力,定义为:
\[ G = H - TS \]
自由能变 (\(\Delta G\)) 可用于判断反应在一定条件下的自发性。如果 \(\Delta G < 0\),反应是自发的;如果 \(\Delta G = 0\),反应处于平衡状态。
### 2.3.2 化学反应的热力学分析
通过对反应物和产物的自由能进行计算,我们可以了解在不同条件下的反应自发性。举例来说,通过下面的吉布斯自由能方程:
\[ \Delta G = \Delta H - T\Delta S \]
我们能够评估在不同温度条件下反应的方向。如果 \(\Delta H\) 和 \(\Delta S\) 的符号相反,反应的自发性会随着温度的改变而改变。在实际应用中,通常需要借助热力学软件或数据库进行精准计算。
通过上述内容,我们不仅理解了化学反应模拟的基础理论,还掌握了一系列模拟技术的基本概念。下文中,我们将深入探讨如何在Star CCM+软件中实际应用这些理论知识。
# 3. Star CCM+软件入门
## 3.1 Star CCM+软件界面和功能概览
Star CCM+是业界领先的计算流体动力学(CFD)软件,广泛应用于工程设计和研发。此软件能够处理复杂流体流动、热传递、化学反应等过程的多物理场耦合模拟。其用户友好的界面设计让工程师即使面对高度复杂的问题,也能够快速搭建模型并执行模拟。
### 3.1.1 用户界面的布局与操作
Star CCM+的用户界面布局十分直观,顶部是菜单栏,提供快速访问常用功能的选项,包括文件管理、模拟设置、结果分析等。界面中央是工作区域,左边是项目浏览器,展示模型树和部件,中间是场景视图,用于可视化模型。右边是属性编辑器,用于修改选中对象的详细属性。
为了熟悉软件操作,可以通过以下步骤开始:
1. 创建新项目:在文件菜单中选择新建项目,输入项目名称并选择保存位置。
2. 导入几何模型:通过导入工具将几何文件(如STL、STEP格式)载入Star CCM+。
3. 设置物理模型:在物理模型菜单中定义模拟的物理条件,比如流体类型、边界条件等。
### 3.1.2 常用模块的介绍和设置
Star CCM+提供了多种模块以满足不同的模拟需求。比如,化学反应模块可以帮助用户模拟和分析化学反应过程,而多相流模块则处理气液固三相流问题。下面简单介绍几个常用模块及其设置:
#### 化学反应模块
- 这个模块允许用户模拟包括燃烧、催化反应在内的化学反应过程。
- 在该模块设置中,需要指定反应物、产物以及反应速率方程。
- 通常,用户需要使用软件内置的数据库或者自定义反应机理。
#### 网格模块
- 网格是CFD模拟的基础,用于划分计算域。
- Star CCM+支持多种网格类型,包括结构网格、非结构网格和多面体网格。
- 网格质量直接影响模拟结果的准确性,因此需要仔细调整网格密度和类型。
#### 计算模块
- 计算模块涉及求解器的选择和控制参数的设置。
- 用户可选择求解器类型(例如压力基或密度基),并根据模型的特性调整时间步长和迭代次数。
#### 结果和可视化模块
- 模拟完成后,需要对结果进行分析。
- Star CCM+提供丰富的后处理工具,如切面分析、矢量图、等值线图等。
- 可视化工具可以帮助用户直观地理解流动和反应过程。
通过以上步骤和模块设置,我们可以看到Star CCM+在模拟化学反应方面具有一整套完善的功能。下面章节将介绍如何使用Star CCM+构建基础化学

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏目录
最低0.47元/天 解锁专栏
买1年送1年
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
MODTRAN 5与GIS融合:地理信息系统中的高级应用案例
参考资源链接:[MODTRAN 5.2.1用户手册:参数设置详解与更新介绍](https://wenku.csdn.net/doc/15be08sqot?spm=1055.2635.3001.10343)
# 1. MODTRAN 5与GIS融合的理论基础
MODTRAN 5(Moderate Resolution Atmospheric Transmittance and Radiance
原子云平台API文档自动化:提高效率与质量的策略
参考资源链接:[原子云平台V1.2 API文档:HTTPS与WebSocket接口详解](https://wenku.csdn.net/doc/85m2syb3xf?spm=1055.2635.3001.10343)
# 1. 原子云平台API文档的重要性
API(Application Programming Interface)文档是IT开发和维护过程中不可或缺的一部分,尤其在服务化和微服务架构日益流行的今天。文档不仅指导
【PSIM射频与微波设计】:无线通信电路仿真探索之旅
参考资源链接:[PSIM初学者指南:使用简单示例操作直流电源与元件连接](https://wenku.csdn.net/doc/644b881ffcc5391368e5f079?spm=1055.2635.3001.10343)
# 1. 无线通信基础与PSIM软件概览
## 1.1 无线通信的发展与现状
无线通信技术是现代社会不可或缺的基础设施,其发展从第一代(1G)的模拟通信到今天的第五代(5G)的高速宽带通信,
确保数据完整性:基恩士上位机TCP协议深入探讨
参考资源链接:[基恩士上位机TCP通信协议详解及应用](https://wenku.csdn.net/doc/6412b711be7fbd1778d48f8e?spm=1055.2635
TIA博途负载均衡技术:自动化系统性能优化的实战攻略
参考资源链接:[优化技巧:解决Win10/Win11下西门子TIA博途运行卡顿问题](https://wenku.csdn.net/doc/37qz7z17es?spm=1055.2635.3001.10343)
# 1. TIA博途负载均衡技术概述
在现代自动化领域,TIA博途(Totally In
GrblController问题诊断与解决:故障排除的快速解决方案

参考资源链接:[GrblController安装与使用教程](https://wenku.csdn.net/doc/6412b792be7fbd1778d4ac76?spm=1055.2635.3001.10343)
# 1. GrblController故障排除概述
## 1.1 故障排除的重要性和目的
故障排除在任何技术系统中都是关键环节,对于Grbl
多物理场仿真
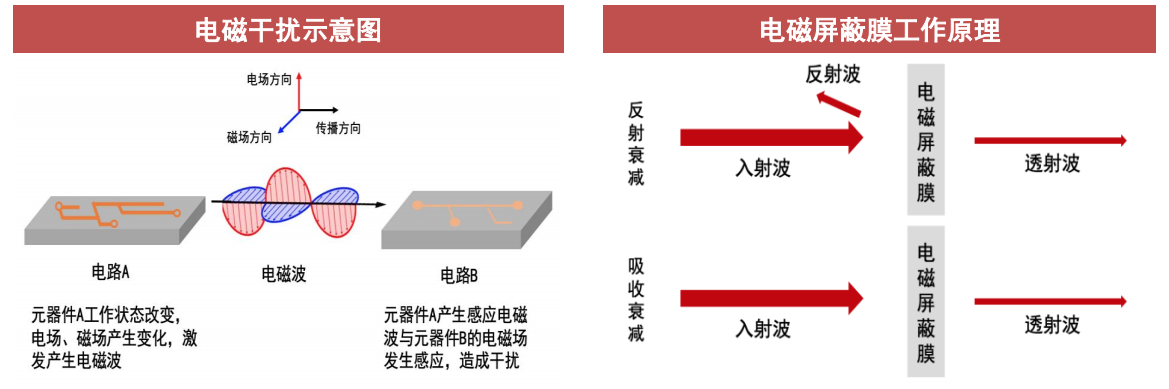
参考资源链接:[cst屏蔽机箱完整算例-电磁兼容.pdf](https://wenku.csdn.net/doc/64606f805928463033adf7db?spm=1055.2635.3001.10343)
# 1. 多物理场仿真的基础概念
在现代工程和科学研究中,多物理场仿真已经成为理解和预测复杂系统行为的重要工具。多物理场仿真涉及至少两个物理场的相互作用,如热力、电磁、
【高级筛选技巧】:Excel中英文菜单对照与高级筛选技巧教程
参考资源链接:[2010版Word与Excel菜单栏功能中英对照](https://wenku.csdn.net/doc/6412b782be7fbd1778d4a8eb?spm=1055.2635.3001.10343)
# 1. Excel高级筛选基础
Excel是数据处理和分析的强大工具,高级筛选是其功能之一,可以让我们在处理大量数据时,迅速找到符合特
【Hillstone SNMP命令行】:提升效率的关键操作指南

参考资源链接:[Hillstone网络设备SNMP配置全攻略](https://wenku.csdn.net/doc/6412b72cbe7fbd1778d49587?spm=1055.2635.3001.10343)
# 1. Hillstone SNMP命令行概述
在现代网络管理中,Hillstone SNMP命令行工具为网络管理员提供了一种高效、灵活的方式来监控和管理网络设备。简单网络管理协议(
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送1年
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )