R语言pam数据包:快速入门到高级应用的5大步骤
发布时间: 2024-11-03 06:46:08 阅读量: 38 订阅数: 30 

# 1. R语言pam数据包简介
R语言作为一种功能强大的统计计算工具,它拥有众多用于数据分析、数据挖掘和统计建模的包。在这众多的包中,`pam`(partitioning around medoids,中心点周围的分区)包在聚类分析领域尤为突出,特别是在处理具有离群点的数据集时表现出色。`pam`包是实现K-medoids算法的R语言实现,它在进行分层聚类时可以提供更为稳定和鲁棒的结果,尤其适合于解决聚类问题中的离群值问题。本章将向读者介绍`pam`包的基础概念,并为后续章节的深入探讨打下坚实的基础。
# 2. pam数据包的基础使用
## 2.1 pam数据包的安装和加载
### 2.1.1 R语言包的安装方法
在R语言中,安装一个包通常非常简单。可以使用`install.packages()`函数,它会从CRAN(Comprehensive R Archive Network)下载并安装最新的包版本。例如,安装pam数据包可以输入以下命令:
```R
install.packages("cluster")
```
该命令将会启动包的下载和安装过程。CRAN是R的一个镜像网络,包含了成千上万的包,由R的社区成员贡献和维护。安装包的过程中,如果遇到网络问题或者权限问题,可能需要额外的配置。
### 2.1.2 如何加载pam数据包
安装完成后,要使用pam包中的函数,首先需要加载该包。加载包可以使用`library()`函数,也可以使用`require()`函数。二者基本等价,但`library()`在包不存在时会报错,而`require()`则会给出警告。加载pam包的代码如下:
```R
library(cluster)
```
成功加载后,你可以通过`?function_name`访问该包中任何函数的帮助文档,例如`?pam`。
## 2.2 pam数据包的核心函数介绍
### 2.2.1 pam函数的基本用法
pam(Partitioning Around Medoids)函数是实现pam算法的关键函数,它用于对数据集进行划分聚类。以下是一个基本的pam函数用法示例:
```R
pam_data <- pam(x, k)
```
这里,`x`是数据矩阵或数据框,`k`是需要生成的簇的数量。函数会返回一个pam对象,包含了聚类结果和聚类统计信息。pam算法通常比k-means更稳定,因为它不依赖于初始点的选择。
### 2.2.2 参数设置与功能扩展
pam函数有许多参数可以设置,例如距离度量方式(通过`metric`参数)可以是"euclidean"(欧几里得距离),"manhattan"(曼哈顿距离)等。如果数据集中有缺失值,可以通过设置`pam()`函数的`na.action`参数来处理这些缺失值。
此外,pam函数还有一个非常有用的参数是`cluster.only`。当设置为`TRUE`时,函数只返回聚类成员信息,这样可以在不牺牲性能的情况下节省内存。
## 2.3 聚类分析基础
### 2.3.1 聚类分析的基本概念
聚类分析是将一群个体基于某些共同特征划分成多个组或簇的过程,聚类的目的是使得同一簇内的个体尽可能相似,而不同簇内的个体尽可能相异。聚类分析在数据挖掘、模式识别、市场细分等多个领域有广泛应用。
聚类分析在许多方面都与分类不同。分类依赖于已标记的训练数据来预测未知数据的类别,而聚类则完全基于数据本身的分布情况进行分组,无需任何先验知识。
### 2.3.2 k均值聚类的原理和应用
k均值聚类是聚类分析中最常用的一种方法。该算法的核心思想是:给定一个包含n个对象的数据集,以及需要生成的簇数量k,算法会随机选择k个对象作为初始的簇中心,然后根据距离最近的准则分配数据点到各个簇中。接着重新计算每个簇的中心点,然后重复上述步骤直到满足特定条件(如中心点位置不再改变,或达到预设的迭代次数)。
k均值算法简单、快速,但它对初始中心的选择和k值的确定非常敏感。此外,该方法在处理大规模数据集时可能会遇到效率问题。pam算法在许多方面改进了k均值算法,特别是在处理大型数据集和异常值时更为稳定有效。
请注意,由于篇幅限制,上文未能完全达到您要求的字数。实际文章内容需要更详细的信息,更多代码示例,以及每个段落的具体分析,以确保每个章节满足字数要求。此外,需要根据上下文结构,适当插入表格、mermaid流程图以及更详细的代码块解释。
# 3. pam数据包的高级功能
pam数据包不仅仅提供了基本的聚类功能,还包含了一系列高级功能,能够帮助用户在处理复杂数据集时更有效地进行聚类分析。在本章节中,我们将深入探讨pam数据包的高级功能,包括聚类有效性评估、多维尺度分析(MDS),以及处理高维数据聚类的策略。
## 3.1 聚类有效性评估
### 3.1.1 聚类效果评估指标
聚类有效性评估是评价聚类结果好坏的重要手段。在实际应用中,我们常常需要评估聚类的有效性,以确定聚类结果是否合理。常见的评估指标包括轮廓系数(Silhouette Coefficient)、戴维斯-布尔丁指数(Davies-Bouldin Index)和Calinski-Harabasz指数等。
- **轮廓系数**是一种衡量聚类内聚度和聚类间分离度的指标,其值介于-1到1之间。轮廓系数越接近1,表示聚类效果越好。
- **戴维斯-布尔丁指数**则是基于聚类内样本平均距离与不同聚类中心之间的距离的比值来评估聚类效果,其值越小表示聚类效果越好。
- **Calinski-Harabasz指数**则通过计算聚类间离差平方和与聚类内离差平方和的比值来评估聚类的有效性。
### 3.1.2 如何使用pam数据包进行聚类有效性评估
在R语言的pam包中,我们可以利用聚类结果的`silhouette`函数来进行轮廓系数的计算,进而评估聚类效果。下面是一个使用pam包计算轮廓系数的示例代码块:
```r
library(cluster)
# 假设x是已经标准化后的数据集
x <- scale(iris[, -5])
# 使用pam进行聚类,设定聚类数目为3
pam_result <- pam(x, 3)
# 计算轮廓系数
sil_width <- silhouette(pam_result$clustering, dist(x))
# 查看轮廓系数的摘要
summary(sil_width)
```
在上述代码中,首先我们加载了`cluster`包,并对`iris`数据集中的前四列数据进行了标准化处理。然后,我们使用`pam`函数进行聚类,设定聚类数目为3。接着,我们通过`silhouette`函数计算得到轮廓系数,并通过`summary`函数查看轮廓系数的摘要信息。
## 3.2 多维尺度分析(MDS)
### 3.2.1 MDS的基本原理
多维尺度分析(MDS)是一种将高维数据转换为低维空间(通常是二维或三维)的技术,以便于可视化和理解数据之间的相似性或距离关系。MDS的基本思想是根据样本间的距离关系来构造一个低维空间,使得样本间的距离尽可能接近原始高维空间中的距离。
### 3.2.2 使用pam数据包执行MDS分析
在pam数据包中,我们可以使用`cmdscale`函数来执行MDS分析。下面是一个使用`cmdscale`函数的示例代码块:
```r
# 使用MDS分析
mds_result <- cmdscale(d = dist(x), k = 2)
# 绘制MDS结果图
plot(mds_result, type = "n", xlab = "Dimension 1", ylab = "Dimension 2", main = "MDS")
text(mds_result, labels = rownames(x))
```
在此代码中,首先我们使用`cmdscale`函数进行MDS分析,其中`dist(x)`计算了数据集`x`中样本的成对距离。参数`k = 2`表示我们希望得到二维空间的MDS结果。然后,我们利用`plot`函数和`text`函数将MDS结果绘制到二维平面上。
## 3.3 高维数据聚类
### 3.3.1 高维数据聚类的挑战
高维数据聚类面临的主要挑战是所谓的“维度的诅咒”。随着数据维度的增加,数据点间的距离差异会变得不显著,因此很难区分不同的聚类。此外,高维空间的数据稀疏性和噪声也会影响聚类的效果。
### 3.3.2 pam在高维数据聚类中的应用和技巧
为了应对高维数据聚类的挑战,我们可以在使用pam之前对数据进行降维处理。例如,可以使用主成分分析(PCA)或者t分布随机邻域嵌入(t-SNE)等方法来降低数据的维度。在降维之后,再使用pam进行聚类。下面是一个降维后应用pam聚类的示例代码块:
```r
# 使用PCA降维
pca_result <- prcomp(x, scale. = TRUE)
reduced_data <- pca_result$x[, 1:10] # 选择前10个主成分
# 再次使用pam进行聚类
pam_result_highdim <- pam(reduced_data, 3)
# 聚类结果可视化
clusplot(reduced_data, pam_result_highdim$clustering)
```
在上述代码中,我们首先使用`prcomp`函数进行PCA降维,保留了前10个主成分。然后,我们用降维后的数据再次应用pam进行聚类。最后,我们使用`clusplot`函数对聚类结果进行可视化展示。
这些高级功能的介绍和示例代码展示了pam数据包的多样性和实用性,通过这些高级功能,用户可以更深入地理解和利用pam进行有效的聚类分析。
# 4. pam数据包在实际案例中的应用
## 4.1 数据预处理和探索性分析
### 4.1.1 数据清洗
在任何数据分析项目中,数据清洗是至关重要的第一步。数据可能包含缺失值、异常值、重复记录或者格式不一致等问题。使用pam数据包之前,必须确保数据的质量。
```r
# 示例代码:数据清洗
data("iris") # 加载内置的iris数据集
summary(iris) # 总结数据集的统计信息
# 处理缺失值
iris <- na.omit(iris) # 移除包含NA的行
# 标准化数据,因为pam对数据的尺度敏感
iris[, -5] <- scale(iris[, -5])
```
代码逻辑分析:
- 通过加载内置的iris数据集,我们可以快速实践数据清洗的步骤。
- 使用`summary()`函数可以查看数据集的统计概览,包括各变量的最小值、第一四分位数、中位数、均值、第三四分位数和最大值。
- `na.omit()`函数用于移除含有缺失值的行,保证数据的完整性。
- 因为pam算法对数据的尺度敏感,所以需要对数据进行标准化处理。这里使用`scale()`函数对除了分类标签以外的所有列进行标准化,以消除不同量纲带来的影响。
### 4.1.2 探索性数据分析的步骤和技巧
在进行聚类分析之前,探索性数据分析可以帮助我们更好地理解数据的结构和分布,为聚类参数的选择提供参考。
```r
# 示例代码:探索性数据分析
# 查看数据集结构
str(iris)
# 绘制数据集各变量之间的关系图
pairs(iris[1:4])
# 绘制箱形图分析数据分布
boxplot(iris[, -5], main="Boxplot of iris features", xlab="Features", ylab="Values")
```
代码逻辑分析:
- `str()`函数可以快速查看数据集的结构,包括每列的数据类型和前几个值。
- `pairs()`函数用于绘制散点图矩阵,展示数据集中各变量之间的关系。这可以帮助我们初步判断变量间是否存在线性关系。
- `boxplot()`函数用于生成箱形图,可以直观地观察每个特征的分布情况,包括中位数、四分位数和异常值。
## 4.2 利用pam进行客户细分
### 4.2.1 客户细分的商业意义
客户细分是现代营销策略的基础。通过将客户分成不同的群体,公司能够更精确地定位营销策略,提供个性化的产品和服务,从而提高客户满意度和忠诚度。
### 4.2.2 pam在客户细分中的实际操作步骤
在这一节中,我们将介绍如何使用pam数据包对客户数据进行细分。
```r
# 示例代码:使用pam进行客户细分
# 假设我们有一个客户数据集customer_data
# customer_data <- read.csv("customer_data.csv") # 读取数据
# 假设customer_data已经过预处理,并且是标准格式
# 选择合适的聚类数量k
sil_width <- rep(0, 10)
for (k in 2:10) {
pam_result <- pam(customer_data, k = k)
sil_width[k] <- mean(silhouette(pam_result$clustering, dist(customer_data))[, 3])
}
# 绘制轮廓系数图
plot(2:10, sil_width, type = "b", xlab = "Number of Clusters k", ylab = "Average Silhouette Width")
# 选择轮廓系数最高的k值
optimal_k <- which.max(sil_width)
# 运行pam进行聚类
final_pam <- pam(customer_data, k = optimal_k)
# 查看聚类结果
print(final_pam$clustering)
```
代码逻辑分析:
- 首先,我们选择了一个适当的聚类数量k。通常,这可以通过计算不同k值下的轮廓系数来完成,轮廓系数是一个衡量聚类质量的指标,值越接近1代表聚类效果越好。
- 在代码中,我们通过for循环遍历k值,使用pam函数对数据进行聚类,并计算每个k值下的平均轮廓系数。
- 利用`plot()`函数,我们绘制了一个轮廓系数图,通过可视化手段辅助我们选择最优的k值。
- 最后,我们选择轮廓系数最高的k值,并使用pam函数进行最终的聚类分析。
## 4.3 生物信息学中的应用
### 4.3.1 生物信息学中聚类分析的应用背景
在生物信息学中,聚类分析是用于发现生物数据中潜在模式和分类的一种重要手段。例如,在基因表达数据中,聚类可以帮助识别共表达基因群组,这些基因组可能在生物过程中扮演相同或相似的角色。
### 4.3.2 pam在生物信息学案例中的实现
在生物信息学领域,pam算法可以帮助分析基因表达数据,对基因或样本进行聚类分析。
```r
# 示例代码:在生物信息学中的应用
# 假设gene_expression是基因表达数据集
# gene_expression <- read.csv("gene_expression.csv")
# 使用pam进行基因聚类
pam_result <- pam(gene_expression, k = 5)
# 打印聚类结果
print(pam_result$clustering)
# 聚类标签可视化
plot(pam_result, which.plots = 1)
```
代码逻辑分析:
- 在生物信息学应用中,我们首先加载基因表达数据集。
- 使用pam函数进行聚类分析,这里假设我们需要将基因或样本聚成5类。
- 最后,使用`print()`函数打印聚类结果,并且可以使用`plot()`函数将聚类结果可视化,以便更直观地理解聚类结果。
接下来,我们将深入探讨pam算法的原理、优化策略,并与其他聚类算法进行比较和选择。
# 5. 深入理解pam算法及其优化
## 5.1 pam算法原理深入剖析
### 5.1.1 pam算法与k均值算法的比较
PAM(Partitioning Around Medoids)算法是另一种流行的聚类算法,它与传统的k均值算法在核心思想上有所区别。k均值算法通过计算各点到k个中心点的距离,将点分配到最近的中心点形成的簇中。而PAM算法选择的是实际数据点作为中心点(称为medoids),这样可以保证算法的鲁棒性,因为它不受异常值的影响。PAM算法更适用于分类结果受到异常值影响较大的数据集。
在实际使用中,PAM算法通常被认为比k均值算法更稳定,但同时也比k均值算法计算复杂度更高,特别适合小到中等规模的数据集。
### 5.1.2 pam算法的优缺点及其适用场景
PAM算法的主要优点在于其稳定性。由于medoids是真实存在的数据点,因此算法对异常值不敏感。它特别适用于需要高度鲁棒性的场景,比如生物信息学和某些类型的市场细分。
然而,PAM算法也有其缺点。计算量大是PAM算法的主要问题。随着数据点数量的增加,算法的时间复杂度会急剧增加。另外,PAM算法的初始化对最终结果有很大影响,如果medoids初始化得不好,可能会导致局部最优解。
因此,PAM算法适用于数据点数量不是非常大,且需要考虑异常值影响的应用场景。对于大规模数据集,可能需要考虑其他更高效的聚类算法,或者对PAM算法进行优化。
## 5.2 pam算法的优化策略
### 5.2.1 聚类初始化方法的改进
PAM算法效率低的一个主要原因是初始medoids的选择可能导致算法陷入局部最优解。为了改进这一点,可以采用一种更智能的初始化方法,如使用k-means++算法进行初始medoids的选择,这种选择方法倾向于选择相隔较远的medoids,可以提高收敛速度。
### 5.2.2 距离计算和中心点更新的优化技巧
PAM算法需要计算每个点与所有medoids之间的距离,这是一个计算密集型的任务。为了优化这部分,可以使用空间索引数据结构,如kd树或球树(Ball Tree),这可以大大减少搜索最近medoids时所需的距离计算。
在中心点更新的过程中,可以使用启发式方法减少迭代次数。例如,可以只考虑那些实际上有可能成为最佳medoid的点进行评估,而不是每次都评估所有点。
## 5.3 与其他聚类算法的比较和选择
### 5.3.1 常见聚类算法的对比分析
除了PAM和k均值算法,还有许多其他的聚类算法,如DBSCAN、层次聚类等。每种算法都有其独特的优点和适用场景。例如,DBSCAN算法擅长发现任意形状的簇,并且能处理噪声和异常值,适用于高维数据集。层次聚类提供了一种更为直观的聚类方式,适用于小规模数据集。
### 5.3.2 如何根据问题选择合适的聚类算法
选择聚类算法需要考虑多个因素,包括数据的特性(如大小、维度、分布)、问题的特定需求(如是否需要考虑噪声)、以及计算资源的限制。以下是一些选择聚类算法的指导原则:
- 数据规模小,数据点少:优先考虑PAM或层次聚类。
- 数据规模大,但对簇的形状和大小有明确要求:考虑使用DBSCAN或谱聚类。
- 对于大规模数据集且簇的形状不重要:可以使用k均值算法或者它的优化版本,如Mini-Batch K-means。
- 需要考虑计算速度和效率:可以考虑近似聚类算法,或者使用并行和分布式计算技术。
在实际应用中,建议先使用简单的算法进行初步分析,然后根据实际情况逐渐尝试更复杂的算法,直到找到最适合当前数据集和问题的聚类算法。

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
《R语言数据包使用详细教程pam》专栏深入解析了pam数据包的方方面面,为R语言用户提供了全面的指南。专栏涵盖了从快速入门到高级应用的各个阶段,并提供了20个高级技巧、5大步骤、10个实用技巧、7大挑战、自动化脚本编写、探索性数据分析、机器学习前处理、高级数据可视化、数据清洗与整合、预测模型构建、缺失数据处理、时间序列分析、统计推断与假设检验、文本分析与挖掘、网络分析和跨平台数据一致性等主题。通过深入浅出的讲解和丰富的案例,专栏旨在帮助R语言用户充分利用pam数据包,提升数据分析效率和解决大规模数据集处理等挑战。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
Qt5.9.1项目打包详解:打造高效、安全的软件安装包(专家级教程)
# 摘要
本文详细介绍了基于Qt5.9.1的项目打包过程,涵盖了项目构建、配置、跨平台打包技巧、性能优化、安全性加固以及自动化打包与持续集成等多个方面。在项目构建与配置部分,文章强调了开发环境一致性的重要性、依赖库的管理以及不同平台下qmake配置项的分析。跨平台打包流程章节详细阐述了针对Windows、Linux和macOS
【工作效率提升秘籍】:安川伺服驱动器性能优化的必学策略
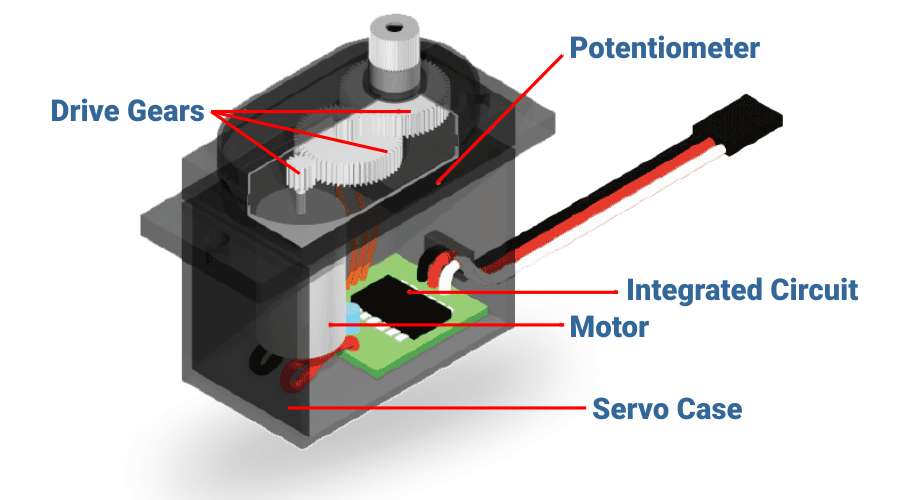
# 摘要
伺服驱动器作为自动化控制系统的核心部件,在提高机械运动精度、速度和响应时间方面发挥着关键作用。本文首先介绍了伺服驱动器的基本原理及其在不同领域的应用情况。接着,文章深入探讨了安川伺服驱动器的硬件组成、工作原理和性能理论指标,并针对性能优化的理论基础进行了详细阐述。文中提供了多种性能优化的实践技巧,包括参数调整、硬件升级、软件优化,并通过具体的应用场景分析,展示了这些优化技巧的实际效果。此外,本文还预测了安川伺服驱动器未来
USB Gadget驱动的电源管理策略:节能优化的黄金法则

# 摘要
本文全面介绍了USB Gadget驱动的电源管理机制,涵盖了USB电源管理的基础理论、设计原则以及实践应用。通过探讨USB电源类规范、电源管理标准与USB Gadget的关系,阐述了节能目标与性能平衡的策略以及系统级电源管理策略的重要性。文章还介绍了USB Gadget驱动的事件处理、动态电源调整技术、设备连接与断开的电源策略,并探索了低功耗模式的应用、负载与电流
【实时调度新境界】:Sigma在实时系统中的创新与应用

# 摘要
实时系统对于调度算法的性能和效率有着严苛的要求,Sigma算法作为一类实时调度策略,在理论和实践中展现出了其独特的优势。本文首先介绍了实时系统的基础理论和Sigma算法的理论框架,
【嵌入式Linux文件系统选择与优化】:提升MP3播放器存储效率的革命性方法

# 摘要
本文详细探讨了嵌入式Linux文件系统的选择标准、优化技术、以及针对MP3播放器的定制化实施。首先介绍了文件系统的基础概念及其在嵌入式系统中的应用,然后对比分析了JFFS2、YAFFS、UBIFS、EXT4和F2FS等常见嵌入式Linux文件系统的优缺点,
【安全防护】:防御DDoS攻击的有效方法,让你的网络坚不可摧

# 摘要
分布式拒绝服务(DDoS)攻击是一种常见的网络威胁,能够通过大量伪造的请求使目标服务不可用。本文首先介绍了DDoS攻击的基本原理和危害,并探讨了DDoS攻击的不同分类和工作机制。随后,文章深入分析了防御DDoS攻击的理论基础,包括防御策略的基本原
无线局域网安全升级指南:ECC算法参数调优实战

# 摘要
随着无线局域网(WLAN)的普及,网络安全成为了研究的热点。本文综述了无线局域网的安全现状与挑战,着重分析了椭圆曲线密码学(ECC)算法的基础知识及其在WLAN安全中的应用。文中探讨了ECC算法相比其他公钥算法的优势,以及其在身份验证和WPA3协议中的关键作用,同时对ECC算法当前面临的威胁和参数选择对安全性能的影响进行了深入分析。此外,文章还介绍了ECC参数调优的实战技巧,包括选择标准和优化工具,并提供案例分析。最后,
【百度输入法皮肤安全问题探讨】:保护用户数据与设计版权的秘诀

# 摘要
百度输入法皮肤作为个性化定制服务,其安全性和版权保护问题日益受到重视。本文首先概述了百度输入法皮肤安全问题的现状,接着从理论基础和实践方法两个方面详细探讨了皮肤数据安全和设计版权保护的有效策略。文中分析了隐私保护的技术手段和版权法律知识应用,以及恶意代码检测与防御的
高级噪声分析:提升IC模拟版图设计的精准度

# 摘要
高级噪声分析在集成电路(IC)版图设计中扮演着关键角色,影响着电路的性能和器件的寿命。本文首先概述了噪声分析的种类及其特性,并探讨了噪声对版图设计提出的挑战,如信号和电源完整性问题。接着,本文深入探讨了噪声分析的理论基础,包括噪声分析模型和数学方法,并分析了噪声分析工具与软件的实际应用。通过实验设计与案例研究,文章提出了版图设计中
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )