
26/10/2017
高通量测序质控及可视化工具包RSeQC
https://mp.weixin.qq.com/s?src=11×tamp=1508994733&ver=475&signature=*3JAynUGvbafOEzRVpHmhXrTGFzDLbZ3vG5TuRnknnXms040Yd7reW…
5/21
divide_bam.py:
随
机
分割
BAM
文
件
(m
个
比
对
结
果
)
为
n
个
文
件
,
每
个
文
件
包
含
m/n
个
比
对
结
果
.
FPKM_count.py:
根
据
read count
和
gene
注
释
文
件
(bed12
格
式
)
计
算
每
个
基
因
的
FPM (fragment per million)
或
者
FPKM (fragment per million mapped reads per kilobase exon).
结
果
类
似
下
面
的
:
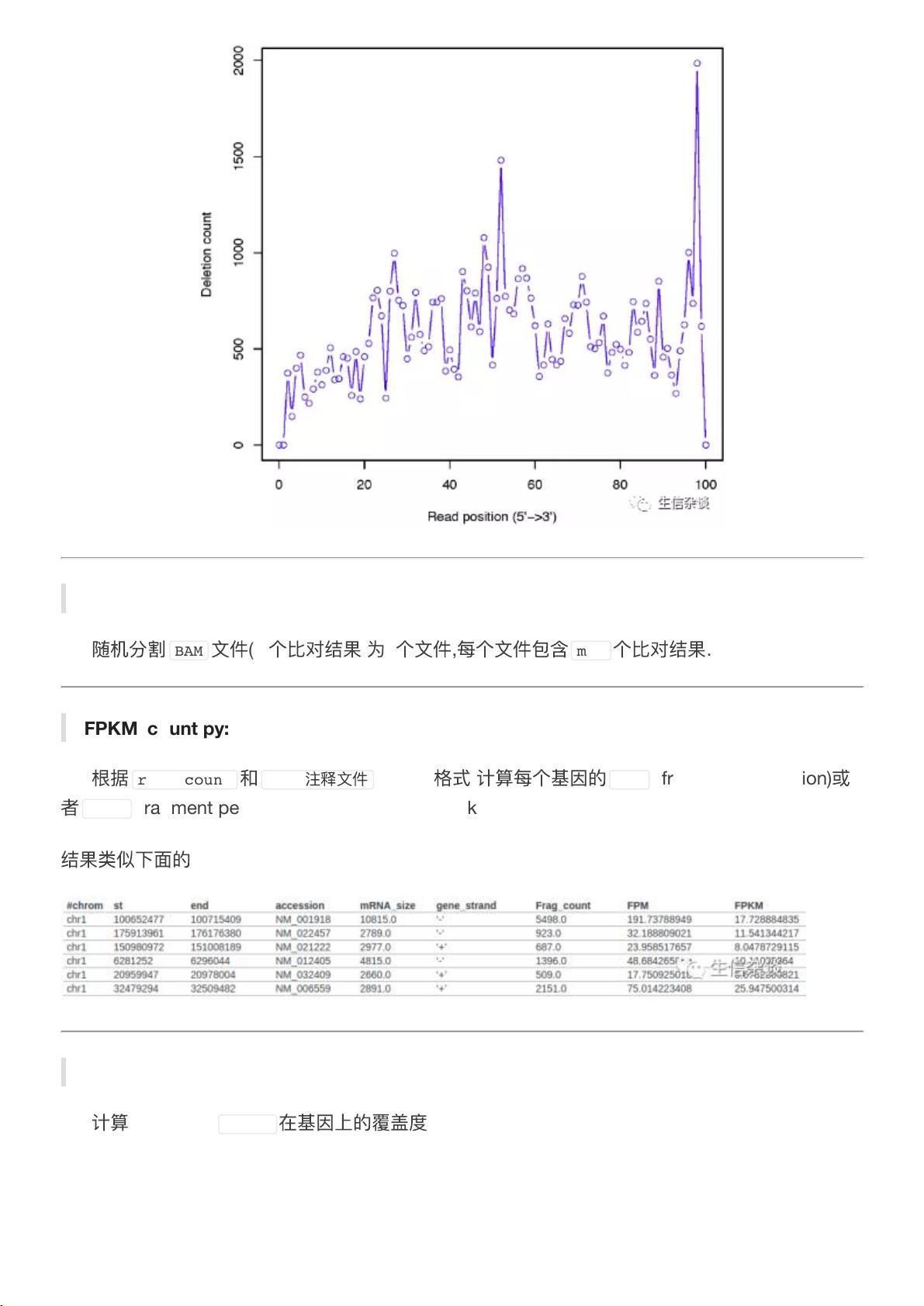
geneBody_coverage.py:
计
算
RNA-seq reads
在
基
因
上
的
覆
盖
度
.
剩余20页未读,继续阅读

不知者无胃口
- 粉丝: 31
- 资源: 328
 我的内容管理
展开
我的内容管理
展开







最新资源
- AirKiss技术详解:无线传递信息与智能家居连接
- Hibernate主键生成策略详解
- 操作系统实验:位示图法管理磁盘空闲空间
- JSON详解:数据交换的主流格式
- Win7安装Ubuntu双系统详细指南
- FPGA内部结构与工作原理探索
- 信用评分模型解析:WOE、IV与ROC
- 使用LVS+Keepalived构建高可用负载均衡集群
- 微信小程序驱动餐饮与服装业创新转型:便捷管理与低成本优势
- 机器学习入门指南:从基础到进阶
- 解决Win7 IIS配置错误500.22与0x80070032
- SQL-DFS:优化HDFS小文件存储的解决方案
- Hadoop、Hbase、Spark环境部署与主机配置详解
- Kisso:加密会话Cookie实现的单点登录SSO
- OpenCV读取与拼接多幅图像教程
- QT实战:轻松生成与解析JSON数据
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


