Application of Matrix Transposition in Bioinformatics: A Powerful Tool for Analyzing Gene Sequences and Protein Structures
发布时间: 2024-09-13 22:13:24 阅读量: 28 订阅数: 25 
# 1. Theoretical Foundations of Transposed Matrices
A transposed matrix is a special kind of matrix in which elements are symmetrically distributed along the main diagonal. It has extensive applications in mathematics and computer science, especially in the field of bioinformatics.
The mathematical definition of a transposed matrix is as follows:
```
A^T = [a_{ij}^T] = [a_{ji}]
```
Here, A is an m x n matrix, and A^T is its transposed matrix.
Transposed matrices have the following properties:
* The number of rows in a transposed matrix equals the number of columns in the original matrix, and vice versa.
* The elements on the main diagonal of a transposed matrix remain unchanged.
* The transposed matrix of a transposed matrix equals the original matrix.
# 2. Applications of Transposed Matrices in Gene Sequence Analysis
Transposed matrices play a crucial role in gene sequence analysis, with applications mainly in sequence alignment and genome assembly.
**2.1 Role of Transposed Matrices in Sequence Alignment**
Sequence alignment is the process of comparing the similarity between two or more sequences, widely used in gene sequence analysis for tasks such as sequence annotation, evolutionary analysis, and gene function prediction. The transposed matrix is central to sequence alignment algorithms, defining the similarity scores between different base pairings.
**2.1.1 Sequence Alignment Algorithms**
Common sequence alignment algorithms include global alignment (Needleman-Wunsch algorithm) and local alignment (Smith-Waterman algorithm). These algorithms are essentially dynamic programming problems, calculating the optimal alignment between two sequences by constructing a score matrix.
**2.1.2 Calculating Weights in Transposed Matrices**
We***mon methods include:
- **PAM matrices:** Evolutionary models based on amino acid sequences, considering the probabilities of point mutations and conserved substitutions.
- **BLOSUM matrices:** Evolutionary models based on protein sequences, considering sequence conservation and the biochemical properties of amino acids.
**2.2 Applications of Transposed Matrices in Genome Assembly**
Genome assembly is the process of assembling short sequence fragments (reads) into a complete genome. Transposed matrices are used in genome assembly to evaluate the overlap regions between reads to determine the optimal arrangement order.
**2.2.1 Principles of Genome Assembly**
Genome assembly usually involves the following steps:
1. **Read overlap:** Identify overlapping regions between different reads.
2. **Graph construction:** Construct a graph with overlapping regions, where nodes represent reads and edges represent overlapping relationships.
3. **Graph traversal:** Use graph traversal algorithms (such as the Euler path algorithm) to find a path in the graph that represents the optimal assembly order of the genome.
**2.2.2 Optimization in the Assembly Process Using Transposed Matrices**
During genome assembly, transposed matrices are used to evaluate the quality of read overlaps. High-quality overlap regions have higher transposed matrix scores, thereby improving the accuracy of the assembly.
**Code Example:**
```python
import numpy as np
# Define a transposed matrix
trans_matrix = np.array([
[1, -1, -1, -1],
[-1, 1, -1, -1],
[-1, -1, 1, -1],
[-1, -1, -1, 1]
])
# Calculate a score matrix for two sequences
seq1 = "ACGT"
seq2 = "ACGT"
score_matrix = np.zeros((len(seq1) + 1, len(seq2) + 1))
for i in range(1, len(seq1) + 1):
for j in range(1, len(seq2) + 1):
score_matrix[i, j] = trans_matrix[seq1[i-1], seq2[j-1]]
# Build a graph
graph = {}
for i in range(len(seq1)):
for j in range(len(seq2)):
if score_matrix[i+1, j+1] > 0:
if i not in graph:
graph[i] = [j]
else:
graph[i].append(j)
# Euler path algorithm
def euler_path(graph):
path = []
while graph:
current = next(iter(graph))
while current in graph:
path.append(current)
next_node = graph[current].pop()
if not graph[current]:
del graph[current]
current = next_node
return path
# Find the optimal assembly order
assembly = euler_path(graph)
```
**Logical Analysis:**
This code demonstrates the application of transposed matrices in sequence alignment and genome assembly.
- **Sequence Alignment:** The code calculates a score matrix for two sequences, which is based on the weights in the transposed matrix to compute the similarity score for each base pairing.
- **Genome Assembly:** The code constructs a graph representing the overlapping relationships between reads and then uses the Euler path algorithm to find a path in the graph that represents the optimal assembly order of the genome.
# 3.1 Role of Transposed Matrices in Protein Folding Prediction
#### 3.1.1 Principles of Protein Folding
Protein folding is a complex biological process involving the transformation of a protein from its linear amino acid sequence into a specific three-dimensional structure. This structure is crucial for the stability and function of proteins. The principles of protein folding are based on thermodynamic and kinetic factors.
Thermodynamic factors include the interaction of the protein with its surrounding environment. The folding process is driven by the minimization of energy, and the folded state of the protein is the state of lowest energy. Kinetic factors include the rate and pathways of protein folding. Protein folding pathways may involve multiple intermediate states before reaching a stable folded state.
#### 3.1.2 Energy Function in Folding Prediction Using Transposed Matrices
Transposed matrices play a vital role in protein folding prediction. They are used as part of an energy function that evaluates the energy of protein folded states. Energy functions typically consist of the following terms:
- **Bonding energy:** Reflects the energy of covalent bonds between amino acids in the protein.
- **Non-bonding energy:** Reflects the non-covalent interactions between amino acids in the protein, such as hydrogen bonds, van der Waals forces, and hydrophobic interactions.
- **Solvation energy:** Reflects the interaction between the protein and surrounding water molecules.
Transposed matrices are used to calculate non-bonding energy. They provide weights for interactions between amino acid pairs, based on the type, distance, and relative orientation of the amino acids.
```python
def calculate_nonbonded_energy(protein):
"""
Calculate the non-bonding energy of a protein.
Parameters:
protein: Protein sequence.
Returns:
Non-bonding energy.
"""
nonbonded_energy = 0
for i in range(len(protein)):
for j in range(i + 1, len(protein)):
nonbonded_energy += transpose_matrix[protein[i]][protein[j]] * distance_matrix[i][j]
return nonbonded_energy
```
By using transposed matrices, the energy function can consider the complexity of interactions between amino acids and provide a more accurate estimate of the energy of protein folded states.
# 4.1 Applications of Transposed Matrices in Disease Diagnosis
### 4.1.1 Relationship Between Gene Mutations and Diseases
Gene mutations are permanent changes in the gene sequence that can lead to abnormal protein structure or

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
KST Ethernet KRL 22中文版:掌握基础配置的7个关键步骤

# 摘要
本文主要介绍KST Ethernet KRL 22中文版的功能、配置方法、应用案例及维护升级策略。首先概述了KST Ethernet KRL 22的基本概念及其应用场景,然后详细讲解了基础配置,包括网络参数设置、通信协议选择与配置。在高级配置方面,涵盖了安全设置、日志记录和故障诊断的策略。文章接着介绍了KST Ethernet KRL 22在工业自动化、智能建筑和环境监测领域的实际应
Masm32性能优化大揭秘:高级技巧让你的代码飞速运行

# 摘要
本文针对Masm32架构及其性能优化进行了系统性的探讨。首先介绍了Masm32的基础架构和性能优化基础,随后深入分析了汇编语言优化原理,包括指令集优化、算法、循环及分支预测等方面。接着,文章探讨了Masm32高级编程技巧,特别强调了内存访问、并发编程、函数调用的优化方法。实际性能调优案例部分,本文通过图形处理、文件系统和
【ABAP流水号生成秘籍】:掌握两种高效生成流水号的方法,提升系统效率
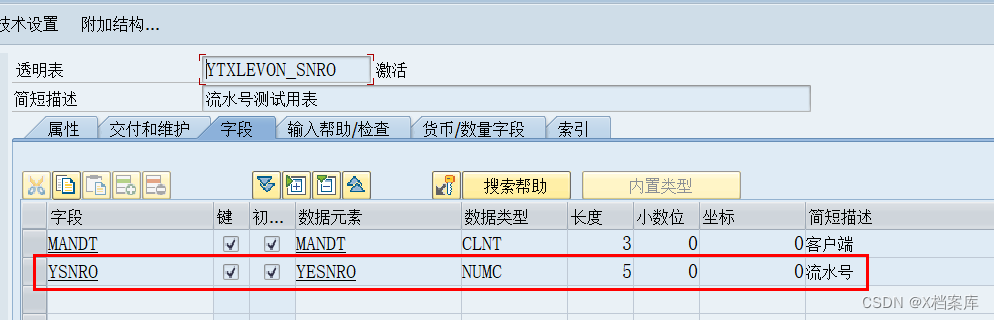
# 摘要
ABAP流水号生成是确保业务流程连续性和数据一致性的关键组成部分。本文首先强调了ABAP流水号生成的重要性,并详细探讨了经典流水号生成方法,包括传统序列号的维护、利用数据库表实现流水号自增和并发控制,以及流水号生成问题的分析与解决策略。随后,本文介绍了高效流水号生成方法的实践应用,涉及内存技术和事件驱动机制,以及多级流水号生成策略的设计与实现。第四章进一步探讨了ABAP流水号
泛微E9流程表单设计与数据集成:无缝连接前后端

# 摘要
本文系统性地介绍了泛微E9流程表单的设计概览、理论基础、实践技巧、数据集成以及进阶应用与优化。首先概述了流程表单的核心概念、作用及设计方法论,然后深入探讨了设计实践技巧,包括界面布局、元素配置、高级功能实现和数据处理。接着,文章详细讲解了流程表单与前后端的数据集成的理论框架和技术手段,并提供实践案例分析。最后,本文探索了提升表单性能与安全性的策略,以及面向未来的技术趋势,如人
TLS 1.2深度剖析:网络安全专家必备的协议原理与优势解读
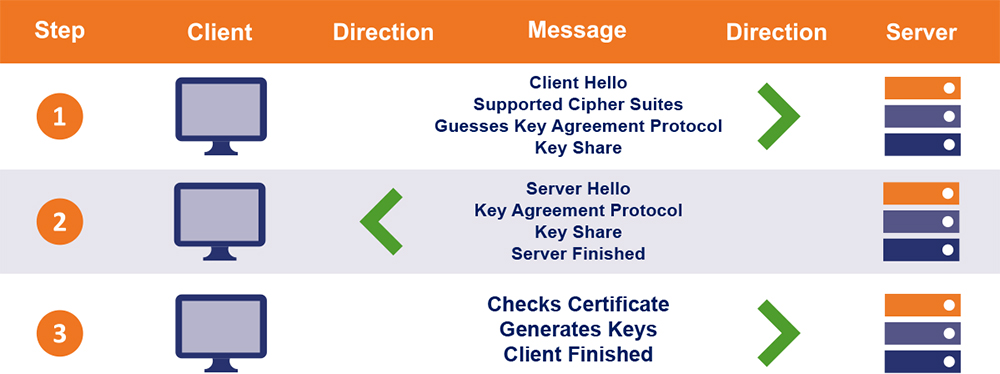
# 摘要
传输层安全性协议(TLS)1.2是互联网安全通信的关键技术,提供数据加密、身份验证和信息完整性保护。本文从TLS 1.2协议概述入手,详细介绍了其核心组件,包括密码套件的运作、证书和身份验证机制、以及TLS握手协议。文章进一步阐述了TLS 1.2的安全优势、性能优化策略以及在不同应用场景中的最佳实践。同时,本文还分析了TLS 1.2所面临的挑战和安全漏
FANUC-0i-MC参数定制化秘籍:打造你的机床性能优化策略
# 摘要
本文对FANUC-0i-MC机床控制器的参数定制化进行了全面探讨,涵盖了参数理论基础、实践操作、案例分析以及问题解决等方面。文章首先概述了FANUC-0i-MC控制器及其参数定制化的基础理论,然后详细介绍了参数定制化的原则、方法以及对机床性能的影响。接下来,本文通过具体的实践操作,阐述了如何在常规和高级应用中调整参数,并讨论了自动化和智能化背景下的参数定制化。案例分析部分则提供了实际操作中遇到问题的诊断与解决策略。最后,文章探讨了参数定制化的未来趋势,强调了安全考虑和个性化参数优化的重要性。通过对机床参数定制化的深入分析,本文旨在为机床操作者和维护人员提供指导和参考,以提升机床性能和
【约束冲突解决方案】:当约束相互碰撞,如何巧妙应对

# 摘要
约束冲突是涉及多个领域,包括商业、技术项目等,引起潜在问题的一个复杂现象。本文从理论上对约束冲突的定义和类型进行探讨,分类阐述了不同来源和影响范围的约束冲突。进一步分析了约束冲突的特性,包括其普遍性与特殊性以及动态变化的性质。通过研究冲突识别与分析的过程和方法,本文提出了冲突解决的基本原则和具体技巧,并通过实践案例分析展示了在商业和技术项目中
提高TIR透镜效率的方法:材料选择与形状优化的终极指南
# 摘要
全内反射(TIR)透镜因其独特的光学性能,在光学系统中扮演着关键角色。本文探讨了TIR透镜效率的重要性,并深入分析了材料选择对透镜性能的影响,包括不同材料的基本特性及其折射率对透镜效率的作用。同时,本文也研究了透镜形状优化的理论与实践,讨论了透镜几何形状与光线路径的关系,以及优化设计的数学模型和算法。在实验方法方面,本文提供了实验设计、测量技术和数据分析的详细流程,
【组态王与PLC通信全攻略】:命令语言在数据交换中的关键作用

# 摘要
随着工业自动化程度的提升,组态王与PLC的通信变得尤为重要。本文首先对组态王与PLC通信进行了总体概述,接着深入探讨了命令语言的基础知识及其在组态王中的具体应用,包括命令语言的定义、语法结构以及数据类型的使用。进一步地,本文分析了命令语言在数据交换过程中的实现策略,包括PLC数据访问机制和组态王与PLC间的数据交换流程。文章还详细讨论了数据交换中遇到的常见问题及解决方法。在此基础上,本文探讨了命令语言的高级应用,并通过实际案例分析了其
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )